 Biomineralisation (½ AC8/9) Biomineralisation (½ AC8/9)
Biomineralisation (½ AC8/9) Biomineralisation (½ AC8/9)1 SWS, nächster Termin noch offen.
Masters-Studiengang Chemie: Eine Hälfte von AC8 (AC als Nebenfach) oder AC9 (AC als Hauptfach)
Während die Untersuchung von Enzymzentren in der Schnittmenge von Biochemie und Koordinationschemie liegt, findet sich in der Schnittmenge von Biochemie und Festkörperchemie/Mineralogie/Materialchemie ein Teilbereich der Bioanorganischen Chemie mit zunehmender Bedeutung: die Biomineralisation. Biominerale sind in der belebten Natur allgegenwärtig, beim Menschen zum Beispiel treten sie in verschiedenen Organen auf:
In den Knochen; dieses Organ enthält einen sich ständig erneuernden Verbund aus dem Protein Kollagen und kristallinem, carbonathaltigem Hydroxylapatit im Mengenverhältnis von ca. 1:2.
In den Otolithen, einzelnen Kristallen von Calcit (CaCO3) in unserem Gleichgewichtsorgan.
Im Speichereisen (Ferritin), aus dem „wenig kristallines“ Eisen(III)-(hydr)oxid bei Bedarf wieder herausgelöst wird.
In dieser Vorlesung wird nach den Bauprinzipien solcher Materialien gefragt. Der Reiz des Themas ist derselbe wie bei der koordinationschemischen Betrachtung von Enzymzentren: die Natur zeigt uns neue Möglichkeiten, Chemie zu betreiben. Bei den Enzymzentren neue Möglichkeiten zum Katalysatordesign, bei den Biomineralen neue Möglichkeiten, Materialien mit ungewöhnlichen Eigenschaften zu entwerfen. In der Vorlesung werden konkret die folgenden Biominerale behandelt:
Bitte beachten Sie die Hinweise auf die Protein Data Bank (PDB) im Skript der Vorlesung Bioanorganische Chemie.
Kristalldaten, bei denen keine Quelle angegeben ist, stammen aus der ICSD.
Informationen zu Raumgruppen finden sich unter der angegebenen Nummer in den International Tables for Crystallography.
Prüfen Sie hier, ob Ihr Browser das Skript korrekt darstellt.
Sie können zum Ausdruck eine pdf-Version verwenden.
Letzte Änderung: 13. Juli 2010.
Biominerale treten in verschieden enger Kombination mit organischen Molekülen auf. Das eine Extrem bilden Verbundmaterialien, bei denen anorganische und organische Komponente in definierter Weise miteinander verwachsen sind und so ein neuartiges Material erzeugen, dessen Eigenschaften von denen der Einzelkomponenten verschieden sind. Das andere Extrem wird durch mehr oder weniger reine anorganische Phasen gebildet, bei denen ein Organismus die Eigenschaften des kristallinen Minerals nutzt. Calciumcarbonat kommt als Baustein in beiden Extremen vor: von den Calcit-Einzelkristallen der Säugetier-Otolithe bis zum Kompositmaterial Permutt, dessen optische Eigenschaften erst im Verbund möglich werden. Dazwischen liegen Coccolithe – kristalliner Calcit, jedoch nicht in Form der üblichen Calcit-Rhomben, sondern als kompliziertes „Werkstück“ ausgebildet.
R 3 c (Nr. 167, hexagonale Aufstellung)
a = 4.984, c = 17.121 Å
6 Ca in 6 b 0, 0, 0
6 C in 6 a 0, 0, ¼
18 O in 18 e 0.2562, 0, ¼
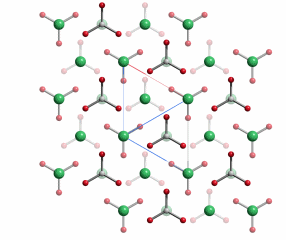
Projektion entlang [0001], 0 ≤ z ≤ ½.
Calcit ist die thermodynamisch stabile Form des Calciumcarbonats. Die Dichte beträgt 2.71 g cm−3. Die Struktur leitet sich von der NaCl-Struktur ab: Ca2+-Ionen bestzen die Plätze der einen Ionensorte, die Carbonat-Ionen die der anderen. Der Einbau der D3h-symmetrischen Carbonat-Ionen ist in geordneter Weise in der NaCl-Struktur nicht möglich – die Lagesymmetrie auf einem Na- oder Cl-Platz ist dort Oh (4/m 3 2/m). Calcit kristallisiert daher nicht in der Kochsalz-Raumgrupe F 4/m 3 2/m, sondern in der trigonalen Untergruppe R 3 2/c, in der alle Carbonat-Ionen dieselbe Ausrichtung ihrer dreizähligen Achsen erfahren.
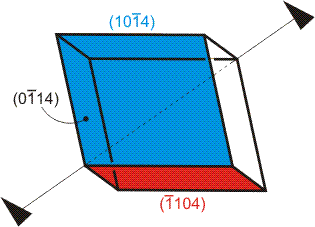
Die Beziehung zur Kochsalz-Struktur ist den typischen Calcit-Rhomboedern deutlich anzusehen. Die ausgebildeten Flächen sind vom Typ (1014). Die oberste Lage einer (1014)-Fläche zeigt die Äquivalenz mit einer NaCl-(100)-Fläche. Wie bei Kochsalz bilden sich also auch bei Calcit elektrostatisch ausgeglichene, unreaktive Oberflächen aus, die Kationen und Anionen im 1:1-Verhältnis aufweisen:
Die charakteristischen Calcit-Rhomben sind von den Flächen (1014), (0114) und (1104) begrenzt. Man beachte die Verwendung des hexagonalen i-Index, i = −h − k:
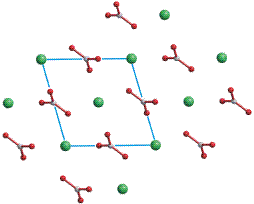
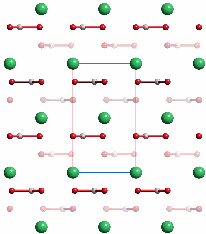
P n m a (Nr. 62)
a = 5.7407, b = 4.9611, c = 7.9672 Å
4 Ca in 4 c 0.24046, ¼, 0.41508
4 C in 4 c 0.08518, ¼, 0.76211
4 O1 in 4 c 0.09557, ¼, 0.92224
8 O2 in 8 d 0.08726 0.47347 0.68065
Projektion entlang [100].
Aragonit ist die zweithäufigste und dichteste Calciumcarbonat-Modifikation (ρ = 2.93 g cm−3). Natürliche Vorkommen dieser schwach instabilen Form (ΔHf0 = +0.12 kJ mol−1 rel. zu 0 für Calcit) sind biogenen Ursprungs. Im Labor lassen sich die typischen Aragonit-Nadeln durch Fällung in homogener Lösung, zum Beispiel durch Carbonatbildung durch Harnstoff-Hydrolyse herstellen [lime1].
P n m a (Nr. 62)
a = 7.15, b = 8.48, c = 4.13 Å; man beachte: a = c × √3
4 Ca in 4 a 0, 0, 0
4 C in 4 c 0.667, ¼, 0.157
4 O1 in 4 c 0.667, ¼, 0.471
8 O2 in 8 d 0.667, 0.118, 0.000
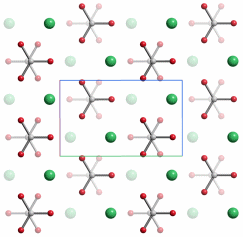
Der metastabile Vaterit, dessen Struktur einen hohen Grad an Lagefehlordnung aufweist, ist die instabilste wasserfreie Calciumcarbonat-Modifikation (ΔHf0 = +3.4 kJ mol−1 rel. zu 0 für Calcit). Vaterit ist ebenso wie das instabile Calciumcarbonat-Monohydrat (Monohydrocalcite) viel weniger verbreitet als Aragonit und Calcit. Im Labor entsteht Vaterit durch schnelle Fällung bei starker Übersättigung, gezielter durch Additive zur Fäll-Lösung wie zum Beispiel l-Aspartat. Biogener Vaterit bildet in der Regel kugelförmige Kristallaggregate („Sphärolithe“), die aus fasrigen, radial verlaufenden Kristallbündeln gebildet werden und deren Durchmesser typischerweise 10 μm beträgt. Vaterit neigt zur Umwandlung in Aragonit oder Calcit, was jedoch durch die Anwesenheit von Carboxylaten oder Phosphat in der überstehenden Lösung verzögert wird. Vaterit gilt als kristalline, aber doch sehr ungeordnete Modifikaiton – was jedoch bei der oft zitierten Aufstellung in der Raumgruppe P b n m nicht zum Ausdruck kommt (P b n m ist die in der Literatur über Vaterit übliche Aufstellung; es handelt sich um eine Nicht-Standardaufstellung von P n m a; bei der oben gegebenen Strukturbeschreibung wurde in die Standardaufstellung transformiert). Die Abbildung zeigt das orthorhombische Strukturmodell in einer Projektion entlang [010] (P b n m) entsprechend [100] (P n m a). Die dreizählige Achse der hexagonalen Modelle verläuft im Bild senkrecht.
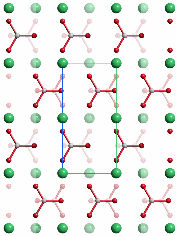
Die hexagonale Symmetrie der Calcium-Anordnung wird in einer Projektion entlang [001] (P b n m) entsprechend [010] (P n m a) deutlich:
Es sind Aufstellungen im hexagonalen Kristallsystem publiziert worden, bei denen die C3-Achse des Carbonats senkrecht auf der C3-Achse des Kristallsystems steht, so dass eine mindestens dreifache Lagefehlordnung resultiert. Insgesamt scheint bei Vaterit noch einiges an Arbeit nötig zu sein.
P 63/m m c (Nr. 194)
a = 4.13, c = 8.49 Å
2 Ca in 2 a 0, 0, 0
2 C in 6 h x, 2x, ¼, x = 0.29
2 O1 in 6 h x, 2x, ¼, x = 0.12
4 O2 in 12 k x, 2x, 0.12, x = 0.38
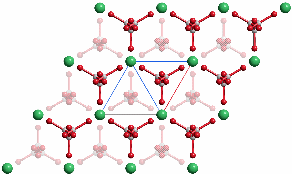
Projektion entlang [0001].
Biogenes amorphes Calciumcarbonat wird wie Vaterit in Form kugelförmiger Aggregate gebildet. EXAFS-Untersuchungen erlauben den Schluss, dass die Nahordnung der Ionen derjenigen in Vaterit nahekommt. ACC ist besser löslich als die kristallinen Modifikationen. Es scheint von zahlreichen Organismen als Vorstufe zu kristallinem Calciumcarbonat oder als Calciumvorrat gebildet zu werden [lime2, lime8].
Otoconien enthalten zahlreiche, wohlausgebildete Einzelkristalle in einer Gelmatrix. Alle vier der genannten Calciumcarbonat-Formen kommen in Organismen vor. Die drei kristallinen Modifikationen werden in Otolithen (wörtlich: „Ohrsteine“) höherer Lebewesen gefunden, während primitivere Organismen zu diesem Zweck Calciumphosphat verwenden. Beim Menschen finden sich die Otolithen im Innenohr. Dort ist das Gleichgewichtsorgan angesiedelt, in dem Nervenzellschichten mit einem aufgelagerten Gel horizontal und vertikal zum Schwerefeld der Erde verlaufen. Das Gel dient als Matrix, in dem wohlausgebildete Calcit-Einkristalle gebildet und zu einem Kristallrasen fixiert werden. Das gesamte Gebilde heißt Otoconia (wörtlich: „Ohrstaub“), in Anspielung auf die große Menge an winzigen Kristallen. Die Formbeständigkeit des Gels ist gerade so hoch, dass die Kristalle an einer ungeregelten Bewegung gehindert werden, dass sie aber sowohl der Gravitation folgen als auch auf Beschleunigungen reagieren können und dabei einen mechanischen Reiz auf die darunterliegenden Nervenenden ausüben können. (Man beachte die unerwartet erfolgreiche Therapie von sogenanntem gutartigen Schwindel, die auf einer Wiedereinpassung einzelner verschobener Kristalle durch gezielte Bewegungen des Kopfes beruht.)
Otoconien, die einen Kristallrasen aus kleinen Kristallindividuen enthalten, kommen auch bei Nicht-Säugern vor, ohne dass eine Beschränkung auf Calcit aufträte. So finden sich im Gleichgewichtsorgan von Reptilien gut ausgebildete Aragonit-Rasen [lime6].
Fische haben wenige große Otolithe mit Zwiebelschalenaufbau. Unter den übrigen Lebewesen sind Otolithen vor allem bei Fischen gut untersucht. Fisch-Otolithen sind erheblich größer und daher auch von größerer träger Masse als die des Menschen, woraus auf eine höhere Empfindlichkeit des Fisch-Gleichgewichtsorgans im Vergleich zu Säugern geschlossen wird. Sie können ohne optische Hilfsmittel leicht aus dem Kopf gängiger Speisefische freipräpariert werden. Während Säugetiere viele Calcit-Kristalle in einer Matrix benutzen, besitzen Knochenfische nur einen großen (Sagitta) zwei kleinere (Astericus und Lapillus) je Ohr. Im Gegensatz zu den wohlausgebildeten Kristallen bei Säugern erinnern Fisch-Otolithe in ihrem Habitus an Kieselsteine. Diese Steine liegen auf Sinneshaarbündeln auf, wodurch die Reizleitung ermöglicht ist. Die vor allem aus Aragonit bestehenden Fisch-Otolithe sind nicht statisch wie die Calcit-Otolithe der Säuger, sondern sie werden allmählich größer, wobei „Tagesringe“ entstehen, die in der Zoologie zur Bestimmung des Alters von Fischen genutzt werden. Dieser schalenförmige Aufbau wirft etwas Licht auf die Otolith-Bildung, zu der drei Bausteine benötigt werden: (1) eine hinreichende Ca2+-Ionen-Konzentration, (2) eine hinreichende Hydrogencarbonat-Konzentration, die durch das Enzym Carboanhydrase sichergestellt wird, und (3) saure Macromoleküle, welche die Keimbildung und das Kristallwachstum steuern und die zum kleineren Teil in die Otolithen eingebaut werden. Die Tagesringe sollen über den Tag variierende Konzentrationen dieser Komponenten widerspiegeln, in dem die Konzentration und damit der Einbau an organischer Matrix in der Abenddämmerung maximal ist und umgekehrt in der Morgendämmerung vor allem Calciumcarbonat abgeschieden wird.
Die Otolithbildung wird durch polymer fixierte COO−-Gruppen gesteuert. Die Einzelheiten der Bildung von Otolithen, also die eigentliche Biomineralisation, sind weitgehend unklar. Sowohl aus Fisch-Otolithen als auch aus Säuger-Otoconium lassen sich saure (Glyco-)Proteine isolieren. Die sauren Eigenschaften sind in einem hohen Anteil an Carboxyl-Funktionen begründet, die beim physiologischen pH-Wert als Carboxylat vorliegen. Träger der Carboxylat-Fragmente sind oxidierte Kohlenhydrat-Bausteine oder Aspartat- und Glutamat-Seitenketten des Proteins. Da Carboxylate Calcium chelatisierend binden können, laufen die derzeitigen Vorstellungen darauf hinaus, in der räumlichen Fixierung von Calcium-Ionen die Ursache für die Nukleation einer bestimmten Modifikation zu sehen.
Die derzeit aktuellsten Arbeiten sind auf Fisch-Otolithe fokussiert. Es scheint so, dass einer der beiden kleinen Steine nicht aus Aragonit, sondern aus Vaterit besteht. Als steuernd werden aspartat- und glutamatreiche Proteine gesehen [lime3]. In Modellierungsstudien gelingt es, zwischen Calcit und Vaterit allein durch den Zusatz von l-Aspartat umzuschalten [lime4].
Die mächtigen Ablagerungen mariner calcifizierender Organismen haben die Erdoberfläche geformt. Die Kalkalpen sind durch Auffaltung biogenen Calciumcarbonats entstanden. Auffällige aragoniterzeugende Populationen wie die Korallenriffe lenken von den gewaltigen Calcit-Mengen ab, die durch marine Einzeller gebildet werden. Kraftmikroskopische Aufnahmen von Calcit-Schilden solcher Organismen belegen, dass diese Einzeller Calcit-Einkristalle zu ihrem Schutz züchten, welche durch dieselben (1014)-Flächen dominiert sind wie unter abiotischen Bedingungen gewachsener Calcit [lime5].
Der kreisförmige Coccolith entwickelt sich aus einer basalen Platte aus organischem Material, die ein ringförmiges, ebenfalls organisches Templat aus Nukleationszentren trägt. Die Calcit-Keimbildung findet an sauren Polysacchariden (PS1 und PS2) statt. Die monomere Einheit von PS2 besteht aus d-Glucuronat, das mit einem 2-meso-Tartratrest ein β-Glycosid bildet. die 3-Position des Tartrats bindet schließlich an Glyoxylat-hydrat. Insgesamt stehen in dieser recht kurzen Wiederholungseinheit vier Carboxylatgruppen zur Verfügung. Die Nukleation führt zu einem Ring von typischerweise zwei Dutzend Calcit-Kristallen, deren C3-Achsen abwechselnd radial (R) und vertikal (V) zum Templatring angeordnet sind:
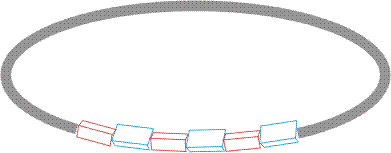
Beim weiteren Wachstum der Kristalle entstehen aus den R-Keimen die nach innen gerichteten, verzahnenden proximale Schilde, während die V-Keime sich zu den von außen wahrnehmbaren distalen Schilden entwickeln.
Muschelschalen bilden – von den Otoconien aus betrachtet – das andere Extrem eines Biominerals. Während die kleinen Calcit- oder Aragonitkristalle der Otoconien wenig organisches Templat enthalten und ihre Eigenschaften wohl nicht der Anwesenheit einer organischen Komponente verdanken, sind Muschelschalen typische Verbundmaterialien. Einer festen Calcit-Außenschale folgt nach innen eine Schicht aus Perlmutt (engl. nacre), aus dem Perlen ausschließlich bestehen. Perlmutt selbst ist ebenfalls ein typisches Komposit-Material. Organische und kristalline Komponente bilden hier einen festen Verbund, dessen Eigenschaften sich von der der einzelnen Komponenten deutlich unterscheidet. Das besondere, von Interferenzfarben geprägte optische Erscheinungsbild verdankt Perlmutt der Anwesenheit von Aragonitscheiben gleicher Dicke, die im Verbund mit proteinbedecktem Chitin vorliegen. Die Proteinschicht führt nicht nur zu höherer mechanischer Stabilität von Perlmutt, sie steuert offensichtlich auch die Nucleation von Aragonit: Aragonit wächst auf frischen Perlmutt-Spaltflächen in geordneter Weise. Wird die Proteinschicht durch Proteinasen zerstört, entfällt die Ordnung bei der Aragonitabscheidung. [lime7] Strukturanalysen an Perlmuttproteinen liegen derzeit noch nicht vor.
Die dirigierenden Eigenschaften carboxylathaltiger Moleküle sind in weitem Umfang genutzt worden, um die Calciumcarbonat-Mineralisation nach dem Vorbild der Natur zu steuern. Die Steuerung geht über die Bevorzugung der gewünschten Modifikation hinaus und schließt die Einstellung der Kristallgestalt ebenso ein wie die Orientierung zum Templat [lime9].
Aragonit-Herstellung (mit Bildern von Aragonit- und Calcit-Kristallen): L. Wang, I. Sondi, E. Matijevic: Preparation of Uniform Needle-Like Aragonite Particles by Homogeneous Precipitation. J. Colloid Interface Sci. 1999, 218, 545–553 [lime1].
ACC: L. Addadi, S. Raz, S. Weiner: Taking Advantage of Disorder: Amorphous Calcium Carbonate and Its Roles in Biomineralization. Adv. Mater. 2003, 15, 959–970 [lime2].
Macromoleküle steuern die Mineralisation: G. Falini, S. Fermani, S. Vanzo, M. Miletic, G. Zaffino: Influence on the Formation of Aragonit or Vaterit by Otolith Macromolecules. Eur. J. Inorg. Chem. 2005, 162–167 [lime3].
Carboxylat-Muster steuern Nukleation und Kristallwachstum: H. Tong, W. Ma, L. Wang, P. Wan, J. Hu, L. Cao: Control over the crystal phase, shape, size and aggregation of calcium carbonate via a l-aspartic acid inducing process. Biomaterials 2004, 25, 3923–3929 [lime4].
Coccolith-Abbildungen: K. Henriksen, S. L. S. Stipp, J. R. Young, P. R. Bown: Tailoring calcite: Nanoscale AFM of coccolith biocrystals. American Mineralogist 2003, 88, 2040–2044 [lime5].
Aragonit in Reptilien-Otoconien: M. Oukda, M. François, H. Membre, A. Bautz, C. Dournon: Crystallographic and chemical composition of otoconia in the salamander Pleurodeles waltl. Hearing Res. 1999, 132, 85–93 [lime6].
Aragonit/Chitin in Perlmutt: S. Blank, M. Arnoldi, S. Khoshnavaz, L. Treccani, M. Kuntz, K. Mann, G. Grathwohl, M. Fritz: The nacre protein perlucin nucleates growth of calcium carbonate crystals. J. Microscopy 2003, 212, 280–291 [lime7].
ACC in den Brustschilden von Läusen: A. Becker, U. Bismayer, M. Epple, H. Fabritius, B. Hasse, J. Shi, A. Ziegler: Structural characterisation of X-ray amorphous calcium carbonate (ACC) in sternal deposits of the crustacea Porcellio scaber. Dalton Trans., 2003, 551–555 [lime8].
Biomimetischer Calcit: Y.-J. Han, L. M. Wysocki, M. S. Thanawala, T. Siegrist, J. Aizenberg: Template-Dependent Morphogenesis of Oriented Calcite Crystals in the Presence of Magnesium Ions. Angew. Chem. 2005, 117, 2–6 [lime9].
Das Skelett ist unserer schwerstes Organ. Osteocyten (Knochenzellen) verteilen sich in einer Matrix aus Kollagen (ca. 25–30 %) und carbonathaltigem Hydroxylapatit (bis 70 %). Weitere Bestandteile der organischen Matrix sind Glycosaminoglycane, die uron-Säuren enthalten und Chondroitinsulfat, ein Polysaccharid, das ebenfalls uron-Säuren enthält. Die Osteocyten reagieren auf mechanischen Reiz durch ständigen Umbau des Knochens. Osteoclasten bauen dabei Knochenbereiche ab, indem durch Kollagenasen die organischen Matrix aufgelöst wird. Durch Carboanhydrase II bereitgestellte Säure löst die Hydroxylapatit-Komponente auf. Osteoblasten füllen die von den Osteoclasten geschlagenen Lücken zuerst mit organischer Matrix, anschließend findet die Calcifizierung statt, bei der das kleine Protein Osteocalcin den Calciumstoffwechsel regelt. Eine im Alter vor allem bei Frauen auftretende Störung des Umbau-Gleichgewichts, die Osteoporose, kann durch Bisphosphonate behandelt werden, deren Wirkung vor allem in einer Hemmung der Osteoclastenaktivität liegt.
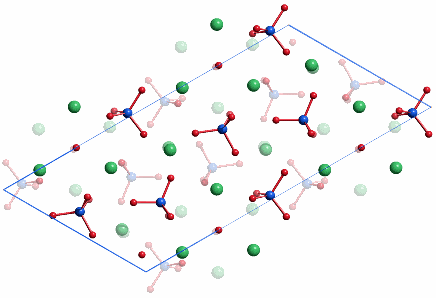
In Knochen kommt sogenannter „biologischer Apatit“ vor, bei dem Ca½PO42−-Äquivalente des Hydroxylapatits teilweise durch HPO42− oder CO32− ersetzt sind. Die Bedeutung von Hydroxid-Ionen im biologischen Apatit ist unklar. Trotzdem kreist die Literatur zu Calciumphosphat-Biomineralen um Hydroxylapatit. Von diesem ist eine monokline Modifikation beschrieben, die jedoch bei Biomineralen nicht diskutiert wird und eine hexagonale Modifikation, über die es eine Fülle aktueller Arbeiten gibt.
P 21/c (Nr. 14)
a = 9.426, b = 6.887, c = 18.856 Å, β = 119.97°;
4 Atome jeder Sorte in der allgemeinen Lage 4 d:
Ca1 0.3430, 0.0024, 0.5870
Ca2 0.3240, 0.4997, 0.5819
Ca3 0.2440, 0.2458, 0.2450
Ca4 0.9920, 0.7540, 0.6231
Ca5 0.2550, 0.7450, 0.3755
P1 0.4010, 0.2480, 0.4330
P2 0.6290, 0.2490, 0.2629
P3 0.0300, 0.7460, 0.4498
O1 0.3300, 0.2400, 0.4920
O2 0.4840, 0.7390, 0.3290
O3 0.1560, 0.2470, 0.5860
O4 0.5870, 0.2650, 0.4810
O5 0.5340, 0.2530, 0.3110
O6 0.1190, 0.7690, 0.5450
O7 0.3590, 0.0680, 0.3800
O8 0.7380, 0.0590, 0.2900
O9 0.0900, 0.5450, 0.4330
O10 0.3380, 0.4220, 0.3750
O11 0.7500, 0.4270, 0.2890
O12 0.0730, 0.9120, 0.4100
O13 0.0000, 0.1924, 0.2450
Projektion entlang [010]:
P 63/m (Nr. 176)
a = 9.352, c = 6.882 Å
4 Ca1 in 4 f 1/3, 2/3, 0.000
6 Ca2 in 6 h 0.240, 0.992, ¼
6 P in 6 h 0.405, 0.371, ¼
6 O1 in 6 h 0.333, 0.478, ¼
6 O2 in 6 h 0.595, 0.467, ¼
12 O3 in 12 i 0.334, 0.257, 0.068
2 O4 in 2 a 0 0 ¼
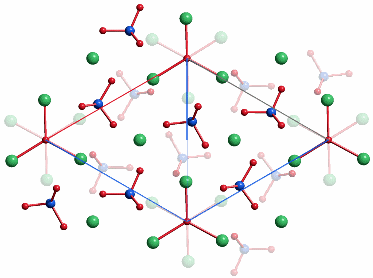
Projektion entlang [0001].
O4 liegt bei diesem Modell auf der Spiegelebene der Raumgruppe P 63/m in der Lagesymmetrie 6, auf gleicher Höhe in [001]-Richtung wie die drei umgebenden Calcium-Ionen der Lagesymmetrie m. Im Fluorapatit ist dies die Lage des Fluor-Atoms. Im heute üblicherweise benutzten Strukturmodell für den Hydroxylapatit ist das Hydroxyl-O-Atom aus der Spiegelebene ausgelenkt und das H-Atom weist von der Spiegelebene weg. Die OH-Lage ist zur Hälfte besetzt, die Lagesymmetrie ist 3 [apatite2]:
2 O4 in 4 e 0 0 0.201
2 H in 4 e 0 0 .0617
Eine Projektion senkrecht zu [001] zeigt diesen Sachverhalt:
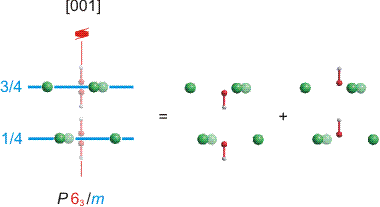
Was dieses über das Volumen eines Kristalls gemittelte Modell in kleineren Volumenabschnitten bedeutet, ist umstritten. Eine Möglichkeit besteht in der Annahme, dass die im mittleren und rechten Teil der Abbildung gezeigte geordnete OH-Verteilung entlang [001] ungestört vorliegt, dass aber keine Ordnung von einem Strang zum nächsten besteht. Eine andere Möglichkeit besteht in der Annahme, dass es bereits in einem Strang zum Richtungswechsel des OH-Vektors kommen kann. Aktuelle Kraftfeldrechnungen favorisieren die zweite Vorstellung [apatite3]. Man beachte bei der Beurteilung dieses auf den ersten Blick unerwarteten Ergebnisses zwei Eigenarten der Hydroxylapatit-Struktur: (1) der Abstand der Hydroxid-Ionen entlang [001] ist mit mehr als 3.4 Å größer als für eine nennenswerte Wasserstoffbrückenbindung erforderlich; (2) bei dem Weg des Protons durch die Spiegelebene, zwischen zwei Calcium-Ionen hindurch, wird es durch Wasserstoffbrückenbindungen zu O-Atomen eines Phosphat-Ions unterstützt.
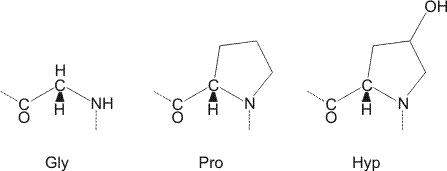
Die ca. 1000 Aminosäuren langen (Gly-X-Y)n-Sequenzen in Typ-I-Kollagen wurden bislang nicht kristallisiert (X meist Pro, Y meist Hyp). Typische, ca. 30 Aminosäuren lange Ausschnitte zeigen immer dasselbe Baumotiv: drei linksgängige (nicht α-helicale) Peptidstränge sind zu einer rechtsgängigen Tripelhelix verwunden. Das Bild zeigt das Trimer des Peptids (Pro-Hyp-Gly)4-Pro-Hyp-Ala-(Pro-Hyp-Gly)5 (PDB-Datei 1CGD):
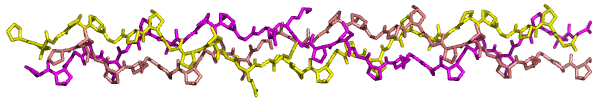
Im Knochen sind die Kollagen-Tripelhelices zu Mikrofibrillen gebündelt. Die Apatit-Komponente ist mit der kristallographischen c-Achse, der [001]-Richtung, parallel zu den Kollagenfasern ausgerichtet.
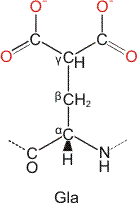
Für die Calcifizierung entscheidend ist das Protein Osteocalcin. Während bei Kollagen typische Calciumbindungsstellen nicht erkennbar sind, sind diese bei Osteocalcin um so augenfälliger. In dem nur 49 Aminosäuren langen Protein sind 3 Glutamat-Seitenketten γ-carboxyliert (siehe Formel), darüberhinaus bilden die drei γ-Carboxyglutamat-Reste (Gla) einen zusammenhängenden Kation-Bindungsbereich.
Dem Strukturbild liegt die PDB-Datei 1Q8H zugrunde. Gezeigt ist die asymmetrische Einheit. Es wird deutlich, dass die drei Calcium-Ionen die potentiellen Calcium-Bindungsstellen nicht absättigen. In [apatite4] ist ein zweites Osteocalcin-Molekül hinzugefügt (das durch die zweizählige Achse der trigonalen Raumgruppe P 3121 erzeugte Molekül). Zusammen bindet das Dimer fünf Calcium-Ionen (eines der drei abgebildeten Ca2+-Ionen liegt in spezieller Lage). Die Autoren untersuchen das Ca5-Muster hinsichtlich seiner Passgenauigkeit mit einer Hydroxylapatit-Oberfläche. Man beachte, dass ein Templateffekt, der von einem Protein mit einem passenden Muster funktioneller Gruppen ausgeht, in fast jedem Teilbereich der Biomineralisation gefordert wird, aber selten nachgewiesen ist. Die Strukturaufklärung an Osteocalcin ist daher ein beachtlicher Fortschritt, der bei anderen Biomineralen aussteht.
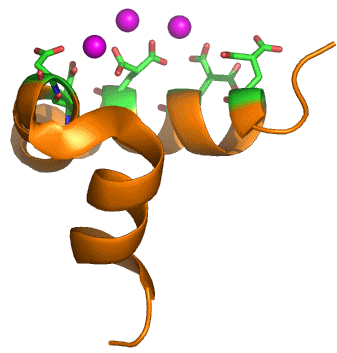
Die Wirkung von Bisphosphonaten als Antiosteoporotica zeigt die Unterschiede zwischen in-vitro- und in-vivo-Experimenten. Die als hydrolysestabilen Derivate des Diphosphats entwickelten Bisphosphonate apatite5 hemmen in vitro sowohl die Bildung als auch den Abbau von Hydroxylapatit. Es liegt nahe, als Ursache die Belegung der Kristalloberfläche durch Bindung der mehrzähnigen Liganden an Calciumzentren anzunehmen – vor allem, wenn man die Analogie zwischen den γ-Carboxy-glutamat-Seitenketten des Osteocalcins und Bisphosphonatstruktur betrachtet. Dieser einfache kristallchemische Vorgang scheint für die in-vivo-Wirksamkeit der Bisphosphonate gegen Osteoporose jedoch belanglos zu sein. Bisphosphonate greifen vielmehr derart in Biosynthesewege ein, dass eine Hemmung der Osteoclastenaktivität resultiert.
Während das Innere eines Zahnes, das Zahnbein (Dentin), dem Knochen sehr ähnlich ist, handelt es sich bei dem außen aufliegenden Zahnschmelz um ein viel dichteres Material. Die chemische Zusammensetzung ist die gleiche wie beim Knochen, die Kristallite sind jedoch bei Enamel deutlich größer. Da Zahnschmelz im Gegensatz zum Knochen nicht lebt, kann sich ein beschädigter oder kariöser Schmelz nicht wieder regenerieren.
Die scheinbar völlig nebensächliche Frage nach der Ausrichtung der Hydroxylgruppe in der Hydroxylapatit-Struktur hat durch ein Experiment zur Biomineralisation Bedeutung erhalten: Wird die Kollagenmatrix eines Knochens von der mineralischen Komponente befreit, und anschließend eine Recalcifizierung mit Calcium und Phosphat versucht, so entsteht Hydroxylapatit in einer neuartigen Morphologie, die bei der freien Kristallbildung nicht beobachtet wird. Fraktales Wachstum einer zu Beginn gebildeten hexagonalen Säule führt zu kugelförmigen Gebilden, für deren Bildung elektrische Felder als maßgeblich diskutiert werden. Die Wechselwirkung mit einem elektrischen Feld setzt jedoch (im Sinne der Symmetrielehre) polare Hydroxylapatit-Domänen voraus, also eine geordnete Ausrichtung der Hydroxylgruppen [apatite7]. Die so gewonnenen Materialien können vielleicht als Zahnschmelzersatz in eine Zahnläsion einwachsen [apatite9].
Biologischer Apatit: S. V. Dorozhkin, M. Epple: Die biologische und medizinische Bedeutung von Calciumphosphaten. Angew. Chem. 2002, 114, 3260–3277 [apatite1].
Hydroxid-Fehlordnung: N. H. de Leeuw: Local ordering of hydroxy groups in hydroxyapatite. Chem. Commun. 2001, 1646–1647 [apatite2].
Kraftfeld-Studie zur Dynamik des Hydroxid-Ions: O. Hochrein, R. Kniep, D. Zahn: Atomistic Simulation Study of the Order/Disorder (Monoclinic to Hexagonal) Phase Transition of Hydroxyapatite. Chem. Mater. 2005, 17, 1978–1981 [apatite3].
Kristallstruktur des Osteocalcins: Q. Q. Hoang, F. Sicheri, A. J. Howard, D. S. C. Yang: Bone recognition mechanism of porcine osteocalcin from crystal structure. Nature 2003, 425, 977–980 [apatite4].
Bisphosphonate: G. K. E. Scriba: Bisphosphonate im Überblick. Pharmazie in unserer Zeit 2000, 29, 50–56 [apatite5].
Wirkmechanismus der Bisphosphonate: G. A. Rodan, A. A. Reszka: Bisphosphonate Mechanism of Action. Current Molecular Medicine 2002, 2, 571–577 [apatite6].
Fraktales Calciumphosphat-Wachstum in Kollagen: S. Busch, H. Dolhaine, A. DuChesne, S. Heinz, O. Hochrein, F. Laeri, O. Podebrad, U. Vietze, T. Weiland, R. Kniep: Biomimetic Morphogenesis of Fluorapatite-Gelatin Composites: Fractal Growth, the Question of Intrinsic Electric Fields, Core/Shell Assemblies, Hollow Spheres and Reorganization of Denatured Collagen. Eur. J. Inorg. Chem. 1999, 1643–1653 [apatite7].
Überblick über die Apatit-Familie: T. J. White, D. Zhi Li: Structural derivation and crystal chemistry of apatites. Acta Crystallogr., Sect. B, 2003, 59, 1–16 [apatite8].
Zahnschmelzersatz: S. Busch: Regeneration von humanem Zahnschmelz. Angew. Chem. 2004, 116, 1452–1455 [apatite9].
Zugvögel scheinen sich am Magnetfeld der Erde orientieren zu können. Bei Brieftauben scheinen sich an der Unterseite des Schnabels Magnetorezeptoren zu finden. Auch scheint in diesen Bereichen Magnetit vorzukommen. Auch in anderen Ordnungen des Tierreichs scheint die Orientierung am Erdmagnetfeld für solche Arten eine Rolle zu spielen, deren Orientierungssinn über lange Strecken funktioniert. Unter den wandernden Fischen ist dies zum Beispiel bei der Forelle sehr wahrscheinlich. Insgesamt jedoch ist der Kenntnisstand zur Biomineralisation von Magnetit oder anderen magnetischen Materialien in Wirbeltieren sehr begrenzt – zu viele Aussagen müssen derzeit noch durch ein „es scheint“ eingeleitet werden. Bis sich dies geändert hat, werden daher in diesem Kapitel ausschließlich die besser untersuchten magnetotaktischen Bakterien vorgestellt.
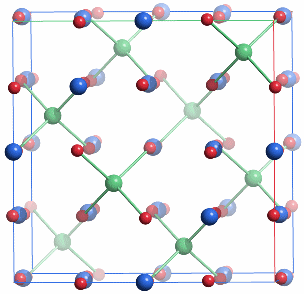
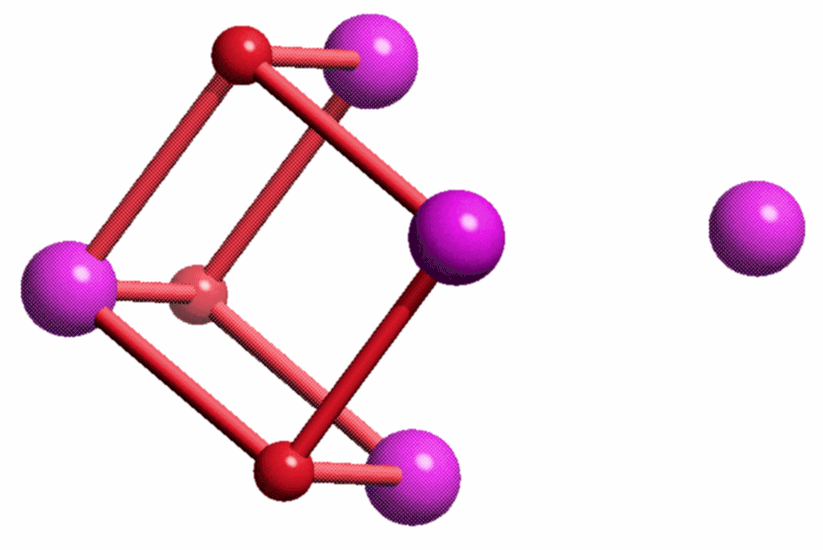
Magnetit ist ein inverser Spinell der Formel FeIII[4t](FeIIFeIII)2[6o]O4, man vergleiche mit dem normalen Spinell MgII[4t]AlIII2[6o]O4, in deren kubisch dichtester Oxid-Packung ein Achtel der Tetraederlücken und die Hälfte der Oktaederlücken gefüllt sind. Der ferrimagnetische Magnetit ist wie Maghämit ein magnetisches Eisenoxid, das technisch in großer Menge hergestellt wird. Die Synthese bedarf nicht der Bedingungen der Festkörperchemie; auch aus wässrigem Medium lässt sich Magnetit gewinnen, indem gefälltes Eisen(iii)-hydroxid, meist wird Ferrihydrit (näheres im Kapitel „Ferritin“) angegeben, mit Eisen(II) umgesetzt wird.
F d 3m (Nr. 227, origin choice 2 = Ursprung in zentrosymmetrischer Lage)
a = 8.392 Å
8 Fe1 in 8 a 1/8, 1/8, 1/8 (tetraedrische Lage A, im Bild grün mit Bindungen)
16 Fe2 in 16 d 1/2, 1/2, 1/2 (oktaedrische Lage B, im Bild blau )
32 O in 32 e x, x, x mit x = 0.2549 (kubisch dichteste Packung, im Bild rot)
Magnetotaktische Bakterien tauchen entlang magnetischer Feldlinien in die Tiefe [magnetite_2004]. Magnetotaktische Bakterien benötigen eine sauerstoffarme Umgebung, wie sie in der aerob-anaeroben Übergangszone in Gewässern, Sedimenten und Böden vorkommen. Magnetotaktische Bakterien richten sich mit Hilfe von Magnetosomen parallel zu den Feldlinien des Erdmagnetfelds aus; sie benutzen Geißeln, um sich entlang der Feldlinien fortzubewegen. Der Sinn der Magnetotaxis ist nicht ganz klar, da auch Mutanten, die keine funktionsfähigen Magnetosomen ausbilden können, anscheinend keinen Wachstumsnachteil erfahren. Dennoch wird Magnetotaxis als Mechanismus gesehen, der den Bakterien das Auffinden von Zonen geringen Sauerstoffgehalts erleichtert.
Biogene Magnetit-Kristalle sind nicht immer isometrisch. Magnetosomen sind Organellen, in denen eine Membran Magnetit-Einzelkristalle oder (111)-Zwillinge umschließt. Die Form der Magnetitkristalle ist nicht nur isometrisch, wie dies im kubischen Kristallsystem bei freiem Wachstum vorgegeben ist, sondern es kommen auch entlang einer der vier [111]-Richtungen gestreckte Kristallindividuen vor. Der Fund gestreckter Magnetit-Kristalle in Sedimenten wird daher als Beleg für deren biogenes Wachstum angesehen (siehe die Diskussion um bakterielles Leben auf dem Mars).
Magnetit-Einzelkristalle sind zu Magnetnadeln arrangiert Die Magnetit-Kristalle sind in langen Reihen angeordnet, so dass Gebilde mit der Funktion von Magnetnadeln entstehen (Figure 1 in [magnetite_2008]). Neben dem üblichen Magnetit wird auch das homologe Sulfid, Fe3S4 (Greigit) gefunden. Die ca 0.1 μm großen Magnetit-Kristalle sind chemisch rein und frei von Protein; sie bilden jeweils eine einzige magnetische Domäne. Die Kette von Magnetosomen verhält sich wie ein einzelner magnetischer Dipol.
Keimbildung und -wachstum finden in einem begrenzten Raum statt. Die Biomineralisation von Magnetit verläuft auf der folgenden Route: Zuerst wird – wahrscheinlich durch Einstülpung der Cytoplasmamembran – die Magnetosommembran gebildet. Anschließend wird Eisen in das Vesikel transportiert, wobei die Aufnahmewege umstritten sind. Die Informationen über Keimbildung und -wachstum sind spärlich. Wie schon bei den Calcium-Mineralen wird die Anwesenheit von Aminosäuren mit Carboxylat-Seitenketten in der Membran vermerkt. Ferner wachsen gleichförmige Magnetitkristalle in vitro in Lösungen, die isolierte Magnetosomenmembran enthalten.
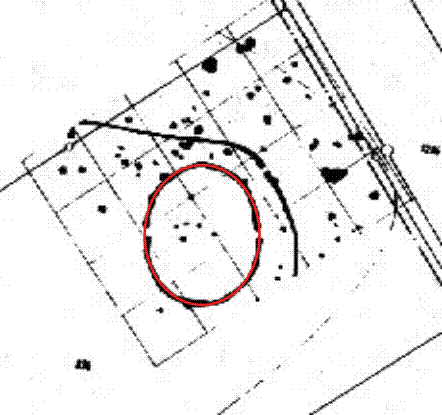
Steinbauten waren nördlich der Alpen in der Jungsteinzeit ebenso unüblich wie im zeitlich anschließenden keltisch-germanischen Kulturbereich. Der übliche Baustoff war Holz, das sich in unserer Witterung in der Regel nicht über historische Zeiträume hält, so dass den zahlreichen Steinbauten der Römerzeit keine erhaltenen Bauwerke aus den vorausgehenden Epochen und wenige aus der nachfolgenden Völkerwanderungszeit gegenüberstehen. Vor allem Holz, das im Kontakt mit dem feuchten Boden steht (Palisaden), wird schnell durch Bakterien und Pilze zersetzt. Magnetotaktische Bakterien, die im Bereich dieser ihrer Nahrungsquelle absterben, reichern den Boden mit biogenem Magnetit an und erzeugen so einen magnetischen „Fingerabdruck“ des ursprünglichen Holzvorkommens. Dies ist eine der Grundlagen der Magnetprospektion in der Archäologie. Das Bild zeigt als Beispiel eine Skizze eines der weltweit ältesten Kalenderbauwerke, der Grabenanlage von Meisternthal im Landkreis Dingolfing-Landau. Die dort durch Magnetprospektion durch den Geophysiker H. Becker untersuchte steinzeitliche Anlage ist nahezu 7000 Jahre alt und damit ca. 2000 Jahre älter als Stonehenge. Die Anlage ist präzise elliptisch (vgl. die einbeschriebene rote Ellipse). Sie zeigt die beiden Sonnenwenden und wahrscheinlich weitere astronomische Ereignisse.
Die Literatur enthält widersprüchliche Meinungen zur Bedeutung aktiver Eisenaufnahmewege in magnetotaktischen Bakterien. Man bedenke die oft reduzierenden Bedingungen in den Lebensräumen dieser Organismen, mit denen die Verfügbarkeit von Eisen(II) einhergeht. Trotzdem werden auch in magnetotaktischen Bakterien Siderophore gefunden, die von den meisten anderen Bakterien zur Sicherstellung des Eisenbedarfs ausgeschieden werden. Generell aquirieren Bakterien in aggresiver Weise das von ihnen benötigte Eisen aus ihrer Umgebung. Die hierzu genutzten zahlreichen (ca. 200 bekannte) „Siderophore“ enthalten Liganden mit hoher Eisen(iii)-Affinität. Meist handelt es sich um Catecholate und Hydroxamate, die in deprotonierter Form extrem stabile Eisen(iii)-Komplexe bilden. Die Siderophore humanpathogener Keime müssen mit dem menschlichen Eisen-Transportprotein Transferrin konkurrieren. Transferrin bildet eine so stabilen Eisen(iii)-Komplex, dass sich die Plasmakonzentratio an freiem Eisen(iii) auf ca. 10−24 m berechnet – was einer einstelligen Zahl an freien Eisen(iii)-Ionen im gesamten Blutplasma entspricht.
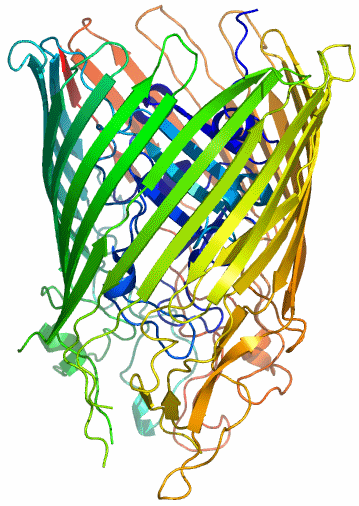
Das am besten untersuchte Siderophor is Enterobactin (Ent), das drei Catecholato(2−)-Liganden zum Aufbau einer FeO6-Umgebung zur Verfügung stellt. FeEnt wird durch FepA, einer Proteinpore in der äußeren Bakterienmembran erkannt und in die Zelle transportiert. Die Abbildung von FepA basiert auf dem PDB-Eintrag 1FEP. Der aus aus ca. 700 Aminosäuren bestehende, lange Proteinstrang bildet nicht nur die Wand der Pore, sondern im Inneren ist ein Verschluss sichtbar, der sich nur für FeEnt öffnet.
Wie erkennt FepA, ob Ent im Inneren ein Eisen-Zentralatom trägt oder ob es leer ist? Die Information hierzu beruht vor allem auf Modellierungsstudien. Bis heute ist FeEnt nicht kristallisiert worden, 1992 wurde jedoch die Struktur des Ent-Vanadium(iv)-Komplexes publiziert, so dass eine Grundlage für die Modellierung vorhanden ist:
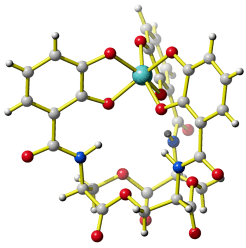
Figure 1 in [magnetite_2003] zeigt eine Abbildung von beladenem und von unbeladenem Ent. In Abwesenheit von Eisen sind die drei auf einem Cyclotriserin-Ring fixierten Brenzcatechin(„Catechol“)-Einheiten nach außen gerichtet. Wasserstoffbrückenbindungen des Typs O(Catechol)–H···O(Amid) halten die eisenchelatisierende Funktion in dieser Stellung. Die Bindung eines Eisen(iii)-Ions führt zur Deprotonierung des Catechols, worauf der Ligand zusammen mit dem Eisen-Ion eine 180°-Drehung in das Siderophor-Innere ausführt und durch eine N–H···O(Catecholat)-Bindung „einrastet“. Das aktive „Einsammeln“ von Eisen-Ionen verlangt den nach außen weisenden potentiellen Chelat-Ligand. Die bei Metalloproteinen übliche Gestaltung einer Metallbindungsstelle aus einzelnen Aminosäure-Seitenketten würde dies nicht leisten können.
Nachdem FeEnt in die Zelle eingeschleust ist, wird das Eisen-Ion durch Zerstörung des Transportmoleküls freigesetzt. Hierzu wird die Amidbindung hydrolysiert.
Um aus einem kubischen Material Magnet-Nadeln zu erzeugen, verkettet die Natur ausgerichtete isometrische Einzelkristalle [magnetite_2005]. Dasselbe Problem stellt sich natürlich auch in der Technik, wo für Speicherzwecke Kristallnadeln aus Magnetit oder Maghämit gewonnen werden. Der Vorgang ist ein Beispiel, wie Natur und Technik auf ganz verschiedenen Wegen dasselbe Ziel erreichen: Goethit, α-FeOOH, ist die thermodynamisch stabilste Phase im System Fe/O/OH. Goethit ist orthorhombisch (Pbnm), in der Kristallstruktur verlaufen kantenverknüpfte FeO6-Oktaeder nur entlang [001]. Es verwundert bei einer derart anisotropen Kristallstruktur nicht, dass Goethit leicht in Form von Nadeln mit großem Länge:Breite-Verhältnis (großem „Aspektverhältnis“) synthetisiert werden kann. Magnetische Eisenoxide werden nun erhalten, indem so schonend dehydratisiert und partiell reduziert wird, dass die ursprüngliche Nadelform erhalten bleibt. Der Vorgang ist eines der vielen Beispiele dafür, wie die belebte Natur ein Funktionsmaterial unter sehr viel milderen Bedingungen herstellen kann als es dies der Technik derzeit möglich ist.
[magnetite_2008]
D. Faivre, D. Schüler:
Magnetotactic Bacteria and Magnetosomes.
Chem. Rev. 2008, 108, 4875–4898.
doi:
10.1021/cr078258w.
[magnetite_2005]
Y. Pan, N. Petersen, A. F. Davilab, L. Zhangb, M. Winklhofer, Q. Liua, M. Hanzlik, R. Zhu:
The detection of bacterial magnetite in recent sediments of Lake Chiemsee (southern Germany).
Earth and Planetary Science Letters 2005, 232, 109–123.
doi:
10.1016/j.epsl.2005.01.006.
[magnetite_2004]
D. A. Bazylinski, R. B. Frankel:
Magnetosome Formation in Prokaryotes.
Nature Reviews Microbiology 2004, 2, 217–230.
doi:
10.1038/nrmicro842.
[magnetite_2003]
K. N. Raymond, E. A. Dertz, S. S. Kim:
Enterobactin: An archetype for microbial iron transport.
PNAS 2003, 100, 3584–3588.
doi:
10.1073/pnas.0630018100.
Mit einer Menge von 3–5 g ist Eisen im menschlichen Körper das wichtigste Übergangsmetall, an das von der Menge her nur noch Zink (ca. 2 g) in etwa heranreicht. Die Hauptmenge von ca. 2/3 ist im Hämoglobin gebunden. Das nächstgrößere Einzelvorkommen ist das Speichereisen, Ferritin, in dem der Körper das Metall für die zahlreichen Funktionen der unterschiedlichsten Eisenzentren vorhält. Der Oxoeisenkern des Ferritins ist neben dem biologischen Apatit der Knochen und Zähne und dem Calciumcarbonat der Otolithen das dritte Biomineral unseres Körpers. In der Literatur wird eine enge Verwandschaft zwischen dem Ferritinkern, der mehr als 4000 Eisenatome umfassen kann, und dem wenig kristallinen Mineral Ferrihydrit gesehen. Der Weg des Eisen von der Nahrung bis in den Ferritinkern ist typisch für ein schwerlösliches Mineral, das überdies einen toxischen Baustein enthält: Mobilisierung durch eine spezialisierte Transportform, strikte Kontrolle der Zustandsform im Körper, Einstellen der notwendigen Oxidationsstufe für die Mineralisation, und schließlich die templatinduzierte Biomineralisation selbst.
Der überragenden Bedeutung des Eisens als Bioelement steht eine äußerst geringe Bioverfügbarkeit gegenüber. Im Verlauf der Evolution reicherte sich die Erdatmospäre mit Sauerstoff an, durch den das an der Boden- oder Gewässeroberfläche vorliegende Eisen in die Oxidationsstufe +III gebracht wurde – aus dem präbiotischen Eisen(II)-sulfid wurde Eisen(III)-(hydr)oxid. Aus dem Löslichkeitsprodukt von maximal ca. 10−38 für frisch gefälltes „Eisen(III)-hydroxid“ errechnet sich bei pH 7 eine Eisen(III)-Konzentration von maximal 10−17 mol L−1.
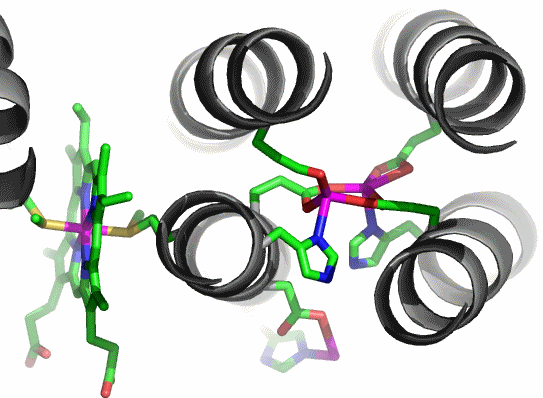
Transferrine binden Eisen(III) mit hoher Bindungskonstante. Das Eisen-Transportprotein im menschlichen Blut ist das Serum-Transferrin (sTf). Die Strukturanalyse zeigt, dass die Natur für den Transport von zwei Eisen(iii)-Atoms ein großes, einsträngiges Protein mit fast 700 Aminosäuren aufbaut, das aus zwei ähnlichen Hälften besteht. Das Bild zeigt die Strukur der N-terminalen Hälfte von humanem sTf (PDB-Code: 1D3K):
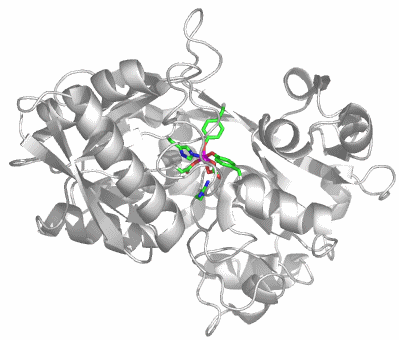
Das aktive Zentrum zeigt die Ursache der CO2-Abhängigkeit des Transferrins:
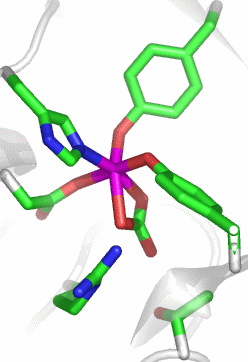
Das FeIII(CO3)-Fragment ist perfekt in das Apoprotein eingebunden. Zwei der sechs Koordinationsstellen des Eisen(iii)-Zentralatoms sind durch einen Carbonato-Liganden belegt. Die hohe Stabilität des Komplexes scheint hiermit nicht im Einklang zu sein, man beachte jedoch die Einbindung des Carbonats in die Gesamtstruktur durch Wasserstoffbrückenbindungen und den weitgehenden Ladungsausgleich. Zwei der Wasserstoffbrückenbindungs-Donoren sind im Bild gezeigt: links unter dem Carbonat-Ion eine kationische Arginin-Seitenkette, die bei physiologischem pH-Wert protoniert vorliegt, außerdem rechts unten die Hydroxylfunktion einer Threonin-Seitenkette. Weitere H-Brücken gehen von Amidfunktionen der Proteinhauptkette aus. Zwei der übrigen Liganden gehören zu den häufigsten metallbindenden Aminosäureseitenketten: Aspartat und Histidin. Die verbleibenden zwei Koordinationsstellen sind demgegenüber durch einen weniger häufigen Liganden besetzt, nämlich durch Tyrosinat.
Wie bei Enterobactin ist die Eisenaufnahme auch bei Transferrin ein Schaltvorgang. Transferrin steht in Konkurrenz zu Siderophoren. Wie Enterobactin ist auch bei Transferrin die Eisenbindung oder -freisetzung durch eine Konformationsänderung optimiert. Bei der Freisetzung öffnet sich der im Bild des Holoenzyms sichtbare Graben oberhalb der Eisenbindungsstelle. Umgekehrt „beißt“ das geöffnete Enzym bei der Bindung eines Eisenatoms an die zuvor mit Carbonat beladene Enzymtasche zu. Die molekulare Ursache des Schaltprozesses ist Gegenstand der Diskussion. Bedeutung wird einem pH-Abfall bei der Bindung an den Transferrin-Rezeptor beigemessen, der die mehrfache Protonierung der Eisenbindungsstelle bewirkt, beginnend mit einer Carbonat-Protonierung. Das Prinzip des Zugreifens zweier beweglicher Proteinteile als Folge eines Schaltsignals wird in [ferritin_2003] näher beleuchtet.
In Ferritinen umschließen Untereinheiten große Hohlräume [ferritin_2010]. Alle Ferritine sind in symmetrischer Weise aus einem strukturell kaum variierten Grundbaustein aufgebaut. Es handelt sich um eine ca. 170 Aminosäuren lange Proteinkette, in der vier α-Helices nahezu parallel zueinander ausgerichtet sind. Bakterien- und Pflanzenferritine sind aus 12 oder 24 gleichartigen Proteinsträngen zusammengesetzt. Im O-symmtrischen (Punktgruppe 432 nach Hermann-Mauguin) 24-mer wird so ein Hohlraum umschlossen, der ca. 8 nm Durchmesser hat. Es existieren keine Strukturanalysen, die sowohl das Apoprotein als auch einen wohlstrukturierten Eisenkern zeigen. Die derzeit publizierten Ferritinstrukturen sind vielmehr im wesentlichen Apoproteinstrukturen, wobei einzelne Metallbindungsstellen sichtbar werden, wenn die intakten Kristalle das Eintauchen in verschiedene metallsalzhaltige Lösungen tolerierten und dabei einige Metallionen eingelagert haben. Säugerferritine bestehen oft aus zwei verschiedenen Proteinsträngen. So sind die 24-mere von menschlichem Ferritin aus H- oder L-Ketten aufgebaut, die Ferroxidase-Zentren aufweisen (H) oder nicht (L). Ein auffallendes Merkmal ist die oft geringe Sequenzhomologie der Ferritine: obwohl die Vier-Helix-Gestalt ubiquitär ist, unterscheiden sich die Aminosäuresequenzen oft erheblich voneinander. Bakterioferritine sind ebenfalls 24-mer. Sie bestehen aus H-Strängen, die außerdem durch Hämeisen-Zentren verknüpft sein können.
Neben diesen „großen“ Ferritinen werden bei Bakterien kleine, dodekamere Ferritine gefunden, welche die Dps-Unterfamilie bilden und deren Kern ca. 500 Eisenatome im ca. 4.5 nm großen Hohlraum enthalten. „Dps“ steht für „DNA-binding protein from starved cells“. Dieser Ferritintyp wird von Bakterien unter Hungerbedingungen oder unter oxidativem Stress produziert, wobei dichte Schichten von Dps-Partikeln im Wechsel mit eingeschlossenen und dadurch geschützten DNA-Schichten gefunden werden. Der Schutz für die DNA ist nicht unspezifisch zum Beispiel im Sinne einer Diffusionsbarriere zu verstehen. Dps oxidiert vielmehr das in Gegenwart von Sauerstoff oder H2O2 toxische Eisen(II) unter H2O2-Verbrauch zu eingelagertem Eisen(iii). Man vergleiche mit den Ferroxidase-Zentren von 24-mer Ferritinen, die Sauerstoff für die Eisen(II)-Oxidation verbrauchen und H2O2 freisetzen.
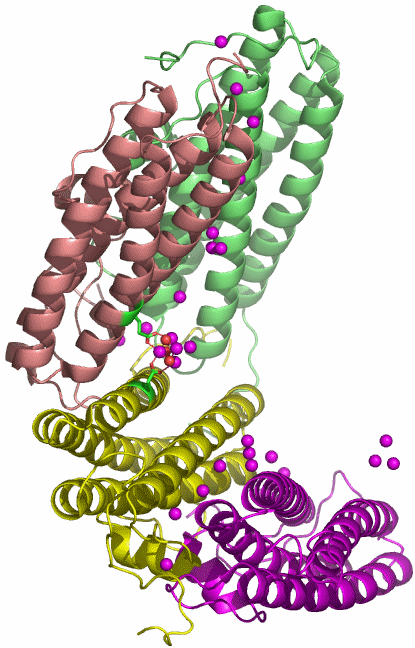
Der PDB-Eintrag 1TKP zeigt die asymmetrische Einheit trigonaler Kristalle von Halobacterium-salinarum-Ferritin in einer Auflösung von 2.2 Å. H. salinarium ist ein halophiles Archäon. Die asymmetrische Einheit des dodekameren Proteins enthält vier Einzelstränge, die in verschiedener Farbe dargestellt sind. Das Tetramer mit den deutlich erkennbaren vier α-Helices eines jeden Einzelstranges ist gebogen und schließt mit zwei weiteren Tetrameren einen T-symmetrischen Hohlraum ein (Punktgruppe 23 nach Hermann-Mauguin, die Symmetrie ist bei 1TKP nicht-kristallographisch). Unter den publizierten Ferritinstrukturen ist 1TKP diejenige, die den Templatcharakter des Proteins am deutlichsten zeigt. Nach Eintauchen der Kristalle in eine Eisen(II)-Lösung unter Luftzutritt gelingt es, den Hohlraum des Dodekamers mit 110 Eisenatomen zu füllen. Die Fe-Atome der asymmetrischen Einheit sind im Bild violett dargestellt; sie finden sich auf der konvexen Seite.
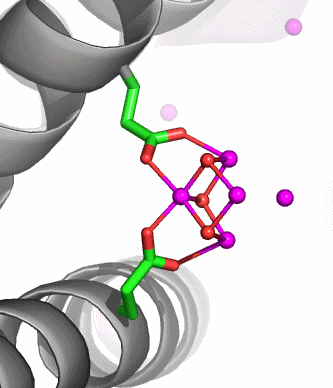
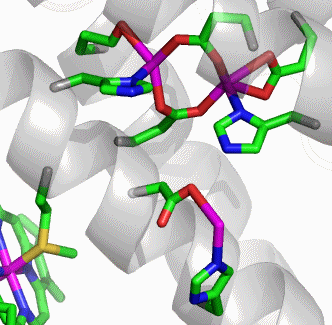
Besonders instruktiv ist die in [ferritin_2004] als Nukleationszentrum NII bezeichnete Position, die im nächsten Bild vergrößert dargestellt ist. Ungefähr waagrecht verläuft eine nichtkristallographische C3-Achse, welche die beiden dargestellten Helices um eine dritte ergänzt, die vor dem Oxoeisen-Fragment liegt und deshalb der Übersicht halber weggelassen wurde. Beachtet man also, dass die beiden dargestellten Glutamat-Reste um einen dritten ergänzt werden, so ergibt sich ein C3-symmetrisches Templat mit insgesamt 6 Haltepunkten. An dieses Templat binden 4 Eisenatome in Form eines Tetraeders derart, dass die drei den Glu-Resten zugewandten Tetraederkanten im geläufigen μ-O1,O2-Bindungsmodus koordinieren. Werden die einzelnen O-Atome im Bild als Hydroxo-Liganden interpretiert, so ergibt sich insgesamt ein Fe4(μ3-OH)3-Motiv. Die Strukturanalyse zeigt nicht, wie das vierte Eisenatom mit dem Rest des Oxoclusters verbunden ist.
Das sich ergebende Muster lässt sich als Ausschnitt aus einer Festkörperstruktur betrachten, wenn Eisen- und Sauerstoff-Atome im Sinne einer anti-NiAs-Struktur auf Arsen- und Nickelplätze gelegt werden, Eisenatome also dicht gepackt werden und Sauerstoffatome in Oktaederlücken gelegt werden:
Bei einem H-Strang liegt in der Mitte zwischen den vier α-Helices ein dinukleares Eisenzentrum. Sowohl das Eisenzentrum selbst als auch dessen Einbau zwischen vier α-Helix-Abschnitte weisen gro0ße Ähnlichkeit mit anderen Dieisenzentren auf. Hierzu gehören Hämerytrhin, Methan-Monoaygenase und Ribonukleotid-Reduktase (RR). (Auf RR wird im Rahmen der Bioanorganischen Chemie nicht eingegangen, da es dort nicht zu einer direkten Metall-Substrat-Wechselwirkung kommt.) Bis heute (2010) konnte kein eisenbeladenes Eukaryonten-Ferroxidase-Zentrum durch Einkristallstrukturanalyse charakterisiert werden. Es liegen jedoch einige Strukturanalysen an Prokaryonten-Zentren vor, bei denen die Beladung des kristallinen Apoproteins mit Eisen gelungen ist.
Über eine Serie von Strukturanalysen an einem metallbeladenem Bakterioferritin nach Kristallisation des Apoproteins und anschließendem Tränken in Metallsalzlösungen wird in [ferritin_2009] berichtet. Im PDB-Eintrag 3E1M ist das erwartete 24-mer beschrieben. Die Abbildung zeigt den typischen 4-Säulenaufbau einer einzelnen Proteinkette. Das Ferroxidasezentrum ist die dinukleare Einheit oben rechts. Die Abbildungen zeigens die Dieisen(II)-Form.
Es zeigen sich zwei Besonderheiten: (1) nur in Bakterioferritinen sind Monomerpaare durch Methionin-Liganden Häm-b-verknüpft (die Funktion dieser low-spin-Zentren ist unklar, eine Bedeutung für die Eisenmobilisierung wird diskutiert). (2) Das auch im folgenden Bild unterhalb des beladenen Ferroxidasezentrums gezeigte Eisenatom, das über Histidin und Aspartat an das Protein gebunden ist, wird als Nukleationszentrum interpretiert. Das Zentrum liegt auf der Innenseite der vom 24-mer gebildeten Hohlkugel.
Das hier gebundene mutmaßliche Eisen(II) könnte vom Ferroxidasezentrum oxidiert werden, um anschließend an einen wachsenden Oxidoeisen(III)-Kern angelagert zu werden. Wird das Ferroxidasezentrum mit Zink beladenen, so wird im Nukleationszentrum ein Eisenatom gefunden, das außer den beiden Aminosäureseitenketten noch Aqualiganden trägt (Figure 10 in [ferritin_2010b]). In Figure 12B derselben Literaturstelle wird ein Modell der Eisenbeladung entworfen, in dem diese Einzelheiten berücksichtigt werden. Die eingelagerten Eisenatome durchlaufen danach keineswegs das Ferroxidase-, sondern das Nukleationszentrum, wo Eisen(II) durch das Ferroxidasezentrum oxidiert wird. Das stets intakte Ferroxidasezentrum ist demnach lediglich der Ort der Sauerstoffreduktion.
Der Ferritinkern gesunder Menschen besteht im wesentlichen aus Ferrihydrit und anderen Eisen(III)-oxid-Phasen, darunter auch Hämatit. Neurodegenerative Erkrankungen wie die Alzheimersche Krankheit oder die progressive supranukleäre (Blick-)Lähmung gehen mit einer verminderten Ferrihydrit-Ablagerung im Hirn-Ferritin einher, zugleich werden vermehrt Eisen(II,III)-oxide gefunden (Magnetit und Wüstit, Fe1−xO, x ca. 0.9) [ferritin_2004b].
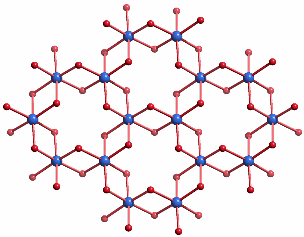
Hämatit kristallisiert in der Korundstruktur. Der charakteristische Baustein sind kantenverknüpfte FeO6-Oktaeder, die ein hexagonales Wabennetz aufbauen:
P b n m (Nichtstandardaufstellung der Raumgruppe Nr. 62 P n m a; P b n m entspricht der Achsenwahl c a b)
a = 4.605, b = 9.960, c = 3.023 Å, alle Atome in 4 c x, y, ¼ mit den folgenden x- und y-Werten:
Fe 0.04770 0.85390
O1 0.70800 0.19960
O2 0.20400 0.05310
H 0.4095 0.0876
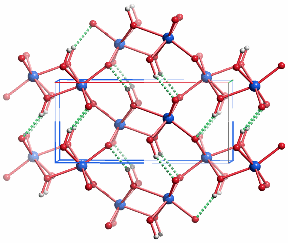
Projektion entlang ca. [001].
In der Goethit-Struktur sind alle Eisen(iii)-Zentren verzerrt oktaedrisch koordiniert. Entlang [001] sind die FeO6-Oktaeder kantenverknüpft, ferner tritt zwischen jeweils zwei Oktaedersträngen paarweise Kantenverknüpfung auf. Die Struktur lässt sich von der Rutil-Struktur durch „kristallographische Scherung“ ableiten, durch welche die Voraussetzung für die Ausbildung von Wasserstoffbrückenbindungen gegeben wird (im bild grün gestrichelt).
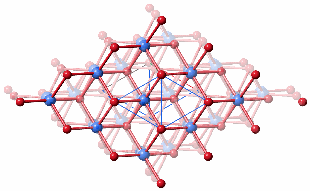
Ferrihydrit ist ein wenig kristallines Eisen(iii)-(hydr)oxid, dessen wesentliches Bauelement in älteren Arbeiten in einer doppelt-hexagonalen Packung von Sauerstoffatomen (Schichtfolge ABAC…) mit Eisen in einem Teil der Oktaederlücken angesehen wird. Die Zusammensetzung wird in [ferritin_2002] mit 5 Fe2O3 · 9 H2O angegeben, entsprechend Fe5O3(OH)9. Auf 12 packungsbildende (Hydr)oxid-Ionen kommen also 5 Eisenatome; die Oktaederlücken sind also ungefähr zur Hälfte (5/12) besetzt.
Das fast amorphe Mineral wird aus zwei Bausteinen zusammengesetzt. So wechseln eine defektreiche Phase in der Raumgruppe P 3 und eine defektfreie Phase mit höherer Symmetrie ab. alle Eisenatome in den defektfreien Domänen sind oktaedrisch durch Oxid oder Hydroxid koordiniert (H-Lagen nicht bestimmt):
P 31c
a = 2.96, c = 9.37 Å
Fe in 4f 1/3, 2/3, z mit z = 0.136
O1 in 2b 0, 0, 0
O2 in 2d 2/3, 1/3, 1/4
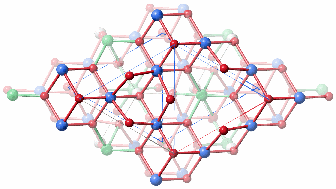
Dieses Grundmotiv wird in einer neueren Arbeit modifiziert [ferritin_2007]. Die Struktur wird hier in einer nichtzentrosymmetrischen, polaren Raumgruppe aufgestellt und mit einer einzigen Elementarzelle beschrieben. Der fast amorphe Charakter des Ferrihydrits wird nun in einer Beschränkung der Domänengröße gesehen (für die kein Grund angegeben wird). Die Zusammensetzung der 2 und 6 nm großen Nanokristallite wird mit Fe5O7(OH) angegeben, also um 4 H2O pro Formeleinheit wasserärmer gesehen:
P 63mc
a = 5.93, c = 9.13 Å
Fe1 in 12d 0.1695, 0.8304, 0.6365
Fe2 in 2b 1/3, 2/3, z mit z = 0.3379
Fe3 in 2b 1/3, 2/3, z mit z = 0.9595
O1 in 2a 0, 0, z mit z = 0.7634
O2 in 2b 1/3, 2/3, z mit z = 0.7634
O3 in 6c x, −x, z mit x = 0.1697, z = 0.2467
O4 in 6c x, −x, z mit x = 0.5227, z = 0.9796
Fe3 ist tetraedrisch koordiniert und in der gezeigten Projektion verdeckt (in der älteren Arbeit liegen tetraedrisch koordinierte Fe-Atome in den Defektblöcken), Fe1 (blau) und Fe2 (grün) sind oktaedrisch koordiniert. Wir diskutieren den Zusammenhang mit der Hämatitstruktur. Zum besseren Vergleich nun die beiden Strukturvorschläge für Ferrihydrit hintereinander, oben die defektfreie Domäne der 2002 publizierten Arbeit, darunter der Vorschlag von 2007:
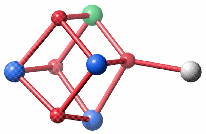
Die unterschiedliche Oktaederlückenfüllung bei den beiden Vorschlägen führt für den jüngeren, nicht aber für den 2002 publizierten, Vorschlag zu Heterokuban-Bausteinen. Werden zudem die tetraedrisch koordinierten Eisenatome berücksichtigt, lässt sich dasselbe Baumotiv herausschälen, das in der eisenreichen Dsp-Struktur gefundenen wurde (Fe1 blau, Fe2 grün, Fe3 grau):
Akaganéit ist ein in den letzten Jahren intensiv untersuchtes Mineral der Hollandit-Familie, A2B8(O,OH)16, A = Ba, K, Pb; B = Fe, Mn; die A- und B-Lage ist oft unterbesetzt. Hollandit ist tetragonal und leitet sich von der Rutilstruktur ab. Die angegebene Formel FeOOH ist idealisiert. Eine realere Formulierung ist zum Beispiel FeO5/6(OH)7/6Cl1/6. Akaganéit wird vor allem dann gefunden, wenn große Ionen wie Cl−, CO32− oder MoO42− im Sinne einer anti-Hollandit-Struktur Kanäle entlang [001]tetragonal besetzen. Möglicherweise kann auch OH− diesen Platz einnehmen. Aktuelle Studien an Akaganéit verfeinern die Struktur nur mit monokliner Symmetrie [ferritin_2003a], [ferritin_2003b]. Das Bild zeigt das Ergebnis der Neutronbeugungsuntersuchung in einer Projektion ungefähr entlang [010]. Die grünen Kreise zeigen die Lagen von Chlorid-ionen an, deren Besetzungsfaktor kleiner als ½ ist. Zum Ladungsausgleich liegt irgendwo in der Struktur die äquivalente Menge an Protonen vor, deren Lage aber auch durch Neutronenbeugung nicht zufriedenstellen bestimmt werden konnte. Werden die nicht zugeordneten Protonen und die Chlorid-Ionen weggelassen, so ist die Zusammensetzung der übrigen Kristallbausteine wie bei Goethit FeOOH.
Das Verknüpfungsmotiv der FeO6-Polyeder ist in den beiden FeOOH-Modifikationen ähnlich. Auch in Akaganéit sind verzerrte FeO6-Oktaeder über Kanten zu Strängen verknüpft. Jeweils zwei Stränge sind durch erneute Kantenverknüpfung miteinander verbunden. Der Unterschied liegt bei der Funktion der Wasserstoffatome. Im Goethit bilden diese Brückenbindungen zu Oxid-Ionen aus, während sie beim Akaganéit in die entlang [010] verlaufenden, aniongefüllten Kanäle hineingerichtet sind.
Ein stärker gegen [010] geneigter Ausschnitt hebt die Kantenverknüpfung der FeO6-Oktaeder entlang [010] hervor:
Das Baumotiv kantenverknüpfter FeO6-Oktaeder durchzieht alle der hier vorgestellten Strukturen. Im Hämatit führt dieses Motiv zu cyclischen Fragmenten. Lineare Stränge kantenverknüpfter Oktaeder, wie sie in Goethit und Akaganéit gefunden werden, sind dagegen in der prototypischen Struktur des Rutils vorgegeben. Die Abbildung zeigt einen entlang [001] projizierten Ausschnitt der tetragonalen AB2-Struktur, der die Verwandtschaft zum Akaganéit besonders hervorhebt:
Nicht nur die beiden Prototypen, Korund und Rutil, sowie die abgeleiteten Festkörperstrukturen sind durch kantenverknüpfte Oktaeder geprägt. Das Baumotiv wird vielmehr auch in mehrkernigen Komplexen dreiwertiger Metalle mit Oxido-Liganden oft gefunden. Die Vermutung ist vernünftig, die oktaedrische Umgebung der Metall-Ionen sowie die Verknüpfung über Ecken und Kanten, nicht jedoch über Flächen, auch in den amorphen Phasen als strukturbestimmend anzunehmen.
Amorphe, ligandstabilisierte Oxidoeisen(iii)-Phasen, deren Struktur den oben zusammengestellten Bauprinzipien folgen sollte, werden seit langem in der Medizin eingesetzt. Sie entstehen beim Neutralisieren saurer Eisen(iii)-Salzlösungen auf pH-Werte zwischen 7 bis ca. 9 anstelle brauner Niederschläge, wenn den Lösungen vor dem Fällen Kohlenhydrate zugegeben wurde. Unter diesen Bedingungen entstehen keine Niederschläge, sondern kaffeebraune Lösungen, die zur Trockne eingeengt werden können und anschließend bei Wasserzugabe wieder dieselben Lösungen ergeben. Lichtstreuversuche ergeben Partikelgrößen von ca. 2 nm. Das Bild zeigt eine solche Lösung, in der Eisen(iii) durch weniger als 20 Mol-% d-Mannit an der Ausfällung gehindert wird:

Bereits in den 40er Jahren des vergangenen Jahrhunderts tauchten Präparationen in den Arzneibüchern auf, die Rohrzucker (Saccharose, engl. sucrose) zu diesem Zweck nutzten. Sie wurden zur Therapie des Eisenmangels oral verwendet. Die Verfügbarkeit des Eisen bei dieser Art der Anwendung ist offensichtlich sehr begrenzt. Eine Renaissance erleben solche Zubereitungen seit der Verfügbarkeit von Erythropoietin (EPO), das vor allem bei der Bekämpfung der Blutarmut von Dialysepatienten eingesetzt wird. Die Therapie mit EPO verlangt hohe Eisengaben, die durch Aufnahme aus der Nahrung nicht zuverlässig zu decken sind. Die nur begrenzte Verfügbarkeit des nanopartikulären Eisens wird nun zum Vorteil. Die zu geringe Resorption auf oralem Wege lässt sich durch parenterale Gabe beheben, die bei diesem Verfahren bei normalen Eisensalzen zu hohe Toxizität tritt bei dem weniger verfügbaren Eisen der Partikel nicht auf. Lediglich die erhöhte Infektionsanfälligkeit durch die Bereitstellung von Eisen bleibt ein Problem (siehe oben).
Sichere Aussagen über die Struktur nanoskaliger Oxoeisen(iii)-Partikel sind nur schwer zu erhalten. Eine neuere Arbeit kommt zu dem Schluss, dass das Baumotiv des Akaganéits vorliegen soll. Ein Vergleich der dieser Aussage zugrunde liegenden Röntgenbeugungsdiagramme (Figure 3 und Figure 4 in [ferritin_2004c] mit dem Pulverdiagramm von kristallinem Akaganéit (Figure 3 in [ferritin_2003a]) und Beugungsdiagrammen von Ferrihydrit (Figure 2 in [ferritin_2002]) zeigt die Unsicherheit solcher Interpretationen.
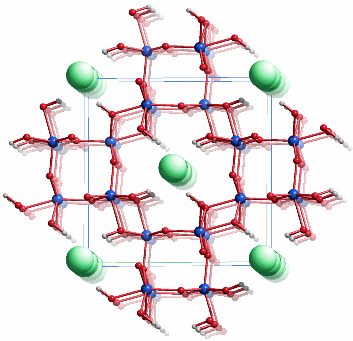
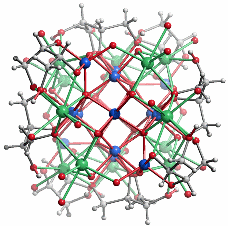
Es gibt nur eine einzige Einkristallstrukturanalyse an einem kohlenhydrat-stabilisierten Oxoeisen(iii)-Nanopartikel. Der Durchmesser von ca. 1.6 nm stimmt mit dem Ergebnis der Lichtstreuung überein, so dass wohl ein typisches Partikel der Lösung kristallisiert werden konnte. Das Übersichtsbild zeigt Kern/Schale-Aufbau (blau: Eisen, grün: Calcium, grau: Kohlenstoff und Wasserstoff, rot: Sauerstoff):
Zehn von insgesamt 14 Eisen(iii)-Zentren bilden einen kompakten Kern, der das Baumuster des Rutils zeigt:
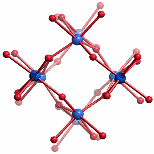
Alternativ lässt sich der innere Kern im Sinne zweier Hämatit-Fragmente interpretieren, die auf symmetrie-unverträgliche Weise ineinandergesteckt sind, so dass weder das Hämatit- noch das Rutil-Baumuster zu einem makroskopischen Kristall weiterwachsen kann; die Abbildung zeigt eines der beiden Hämatit-Fragmente:
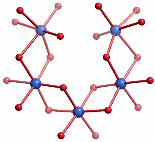
[ferritin_2010]
R. R. Crichton, J.-P. Declercq:
X-ray structures of ferritins and related proteins.
Biochim. Biophys. Acta 2010, 1800, 706–718.
doi:
10.1016/j.bbagen.2010.03.019.
[ferritin_2010a]
F. Bou-Abdallah:
The iron redox and hydrolysis chemistry of the ferritins.
Biochim. Biophys. Acta 2010, 1800, 719–731.
doi:
10.1016/j.bbagen.2010.03.021.
[ferritin_2010b]
N. E. Le Brun, A. Crow, M. E.P. Murphy, A. G. Mauk, G. R. Moore:
Iron core mineralisation in prokaryotic ferritins.
Biochim. Biophys. Acta 2010, 1800, 732–744.
doi:
10.1016/j.bbagen.2010.04.002.
[ferritin_2010c]
E. Chiancone, P. Ceci:
The multifaceted capacity of Dps proteins to combat bacterial stress conditions: Detoxification of iron and hydrogen peroxide and DNA binding.
Biochim. Biophys. Acta 2010, 1800, 798–805.
doi:
10.1016/j.bbagen.2010.01.013.
[ferritin_2009]
A. Crow, T. L. Lawson, A. Lewin, G. R. Moore, N. E. Le Brun:
Structural Basis for Iron Mineralization by Bacterioferritin.
J. Am. Chem. Soc. 2009, 131, 6808–6813.
doi:
10.1021/ja8093444.
[ferritin_2007]
D. R. Strongin, M. A. A. Schoonen, B. L. Phillips, J. B. Parise:
The Structure of Ferrihydrite, a Nanocrystalline Material.
Science 2007, 316, 1726–1729.
doi:
10.1126/science.1142525.
[ferritin_2004]
K. Zeth, S. Offermann, L. O. Essen, D. Oesterhelt:
Iron-oxo clusters biomineralizing on protein surfaces: Structural analysis of Halobacterium salinarum DpsA in its low- and high-iron states.
Proc. Nat. Acad. Sci. USA 2004, 101, 13780–13785.
doi:
10.1073/pnas.0401821101.
[ferritin_2004a]
D. S. Kudasheva, J. Lai, A. Ulman, M. K. Cowman:
Structure of carbohydrate-bound polynuclear iron oxyhydroxide nanoparticles in parenteral formulations.
J. Inorg. Biochem. 2004, 98, 1757–1769.
doi:
10.1016/j.jinorgbio.2004.06.010.
[ferritin_2004b]
C. Quintana, J. M. Cowley, C. Marhic:
Electron nanodiffraction and high-resolution electron microscopy studies of the structure and composition of physiological and pathological ferritin.
J. Struct. Biol. 2004, 147, 166–178.
doi:
10.1016/j.jsb.2004.03.001.
[ferritin_2003]
H. M. Baker, B. F. Anderson, E. N. Baker:
Dealing with iron: Common structural principles in proteins that transport iron and heme.
Proc. Natl. Acad. Sci. 2003, 100, 3579–3583.
doi:
10.1073/pnas.0637295100.
[ferritin_2003a]
J. E. Post, P. J. Heaney, R. B. v. Dreele, J. C. Hanson:
Neutron and temperature-resolved synchrotron X-ray powder diffraction study of akaganéite.
Am. Miner. 2003, 88, 782–788.
doi:
doi not available.
[ferritin_2003b]
K. Ståhl, K. Nielsen, J. Jiang, B. Lebech, J. C. Hanson, P. Norby, J. van Lanschot:
On the akaganéite crystal structure, phase transformations and possible role in post-excavational corrosion of iron artifacts.
Corrosion Science 2003, 45, 2563–2575.
doi:
10.1016/S0010-938X(03)00078-7.
[ferritin_2002]
E. Jansen, A. Kyek, W. Schäfer, U. Schwertmann:
The structure of six-line ferrihydrite.
Appl. Phys. A 2002, 74, S1004–S1006.
doi:
10.1007/s003390101175.
In der belebten Natur kommen amorphe Kieselsäureformen in beträchtlicher Menge vor, während Silicate als Biominerale keine Rolle spielen. Am besten untersucht sind Kieselalgen (Diatomeen und Radiolarien), genetesch gut untersucht sind verkieselnde Schwämme. In beiden Gruppen wurden Proteine isoliert, die mit der Polykondensation von Kieselsäure assoziiert sind – die Silaffine bei den Diatomeen, die Silicateine bei den Schwämmen. Die Verkieselung von Pflanzen wie Schachtelhalmen, vor allem aber den Gräsern, ist seit langem bekannt, es gibt jedoch kaum Erkenntnisse über die Einzelschritte der SiO2-Biomineralisation in Pflanzen.
Kieselalgen (engl. diatoms) besitzen ein aus zwei „Theken“ bestehendes Exoskelett, von dessen beiden Schalen bei der vegetativen Zellteilung jeweils eine an jede Tochterzelle weitergegeben wird und jeweils eine neue kleinere Schale (die „Hypotheka“) synthetisiert wird (Fig. 2 in [silica1]). Aufgrund dieses Mechanismus kommt es zur „Diminution“ – ein Teil der Population wird immer kleiner. Kieselalgen korrigieren diesen Vorgang durch geschlechtliche Vermehrung, bei der Theken in arttypischer Größe vollständig neu gebildet werden.
Die Theken sind nicht dicht, sondern sie erlauben den Stoffaustausch aufgrund ihres hierarchischen, porösen Aufbaus (Fig. 1 in [silica1]). Die Bildung des hierachischen Musters ist auf der mikroskopischen Größenskala in den Grundzügen verstanden: Kieselsäure wird in Vesikeln akkumuliert („silica deposition vesicles“, SDVs), in denen ein nicht näher bekannter „Cofaktor“ die Polykondensation trotz der hohen Si-Konzentration verhindert. Die kugelförmigen Vesikel bilden eine Monolage dichtest gepackter SDVs. In diese Form hinein wird Polykieselsäure ausgegossen. Die Vesikel bilden nun eine Tochtergeneration kleinerer Vesikel innerhalb jeder Wabe aus dem ersten Schritt. Anschließend wiederholt sich der Vorgang so oft mit kleineren Vesikel-Generationen wie die Schale Hierarchiestufen aufweist (Fig. 6 in [silica1]).
Auf biochemischer Ebene gibt es Kenntnisse über kieselsäurekondensierende Proteine, den Silaffinen, während der mit diesen zusammenspielende, polykondensations-verhindernde „Cofaktor“ dagegen bisher nicht bekannt ist.
Silaffine lassen sich aus Kieselalgenschalen durch auflösen der anorganischen Komponente isolieren (Fig. 3 in [silica1]). Silaffine sind posttranslational veränderte Proteine, die reich an Lysin (Lys, Einbuchstabencode: K) und Serin (Ser, S) sind. Durch Phosphorylierung vor allem von Ser, aber auch an γ-hydroxyliertem Lys, sowie durch Methylierung und Aminoalkylierung von Lys entsteht ein zwitterionisches Protein (Fig. 4 in [silica1]). Schonend isoliertes Silaffin-1A (natSil-1A) katalysiert die Kieselsäure-Polykondensation in vitro, wobei sphärische Partikel gebildet werden (Fig. 7 in [silica1]).
Sowohl bei den verschiedenen SiO2-Modifikationen als auch bei den Silicaten ist die tetraedrische Umgebung von Silicium die Regel, von der es nur wenig Ausnahmen gibt. Bei SiO2 ist die Ausnahme die Hochdruckmodifikation Stishovit, der in Einschlagskratern von Meteoriten gefunden wurde und der in der Rutil-Struktur kristallisiert. Es wird daher davon ausgegangen, dass amorphe Kieselsäureformen stets tetraedrisch koordiniertes Silicium enthalten.
Kristalline Silicate haben nach dem derzeitigen Kenntnisstand in Organismen keine Bedeutung. Unter den nichtkristallinen Kieselsäuren haben Opale einen engen Bezug zu biogenem SiO2. Opale haben die Formel SiO2 · x H2O. Unterhalb eines Wassergehaltes von ca. 7 % sind Opale hart.
Zur Beschreibung des Verknüpfungsgrades von Siliciumzentren hat sich eine Nomenklatur eingebürgert, bei der die Zahl der nächsten Silicium-Nachbarn um das betrachtete Atom herum einem „Q“ als Hochzahl angefügt wird: ortho-Kieselsäure ist eine Q0-Spezies, Dikieselsäure enthält zwei Q1-Zentren, in linearer Trikieselsäure liegt die Abfolge Q1-Q2-Q1 vor, Cyclotrikieselsäure enthält nur Q2-Bausteine. Quarz besteht aus Q4-Baueinheiten.
Nach dem derzeitigen Kenntnisstand nutzen Kieselalgen die in natürlichen Wässern gelöste ortho-Kieselsäure, H4SiO4. ortho-Kieselsäure ist eine schwache Säure, deren pKA mit ca. 9–10 angegeben wird. Bei physiologischem pH liegt also die undissoziierte Säure vor. Übersteigt die Konzentration der Kieselsäure die Löslichkeit amorpher kondensierter Formen von ca. 100 ppm entsprechend ca. 1 mm, so beginnt die Polykondensation. Die pKA-Werte der Silanol-Funktionen an den verschiedenen Qn-Spezies hängt vom Kondensationsgrad ab. Q0-, Q1- und Q2-gebundene Hydroxylgruppen sind so schwach sauer, dass sie bei physiologischem pH nicht protolysiert sind. pKA-Werte für Q3-Silanolgruppen sind kleiner als 7. In [Shaw 2005] wird der Q3-pKA bei 4–5 gesehen, der Wert für Q2 bei 8–9. In neutraler Lösung wird die Q3-Bildung durch den Energiegewinn bei der Dissoziation von Q3-Spezies vorangetrieben, während sich oberhalb eines pH-Wertes von ca. 11 die verschieden stark kondensierten Silicate nebeneinander beobachten lassen. Analog lässt sich die Verteilung über zahlreiche Spezies in saurer Lösung beobachten, wo nur nicht-protolysierte Kieselsäuren vorliegen. Der pKA-Wert kondensierter kolloidaler Partikel wird mit wenig unter 7 angegeben.
Die einzelnen Stufen der Polykondensation lassen sich bei Kieselsäuren nicht fassen oder gar geplant synthetisieren wie bei den Polyphosphorsäuren (wie würden Sie gezielt Pentanatriumtriphosphat herstellen?). Strukturinformationen stammen vor allem aus zwei Quellen: (1) In Silicaten können die Anionen der Kieselsäuren isoliert und charakterisiert werden. Die größeren nicht-polymeren Oligosilicat-Ionen geben dabei einen Eindruck vom Aufbau kleiner Polykieselsäurepartikel in einer wässrigen Lösung. Das folgende Bild zeigt als Beispiel ein nur aus Q3-Einheiten aufgebautes Dodekasilicat (nach Liebau ein Doppelsechserring-Silicat), das zu den größten wohldefinerten Teilchen gehört, die durch 29Si-NMR-Spektroskopie in wässrig-alkalischen Lösungen nachweisbar sind. Typisch für Oligo- und Polysilicate ist die ausschließliche Eckenverknüpfung der SiO4-Tetraeder.
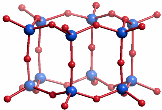
Ein anderes hochsymmetrisches Oligosilicat, das nur aus Q3-Einheiten besteht, ist das Doppelviererring-silicat Si8O208−, das gemäß Si8(μ-O)12(O−)8 würfelförmigen Aufbau hat. (2) Denkt man sich die terminalen O−-Funktionen in dieser Heterocuban-Struktur durch H-Atome ersetzt, so wird ein Silsesquioxan erhalten. Sowohl die Silsesquioxane selbst als auch Derivate, in denen die H-Atome zum Beispiel durch Alkylreste ersetzt sind, sind in großer Zahl isoliert und strukturell untersucht worden. Die erhaltenen Strukturen können als Modelle für Polykieselsäuren angesehen werden. Auch hier ist das Ergebnis, dass die tetraedrischen Baueinheiten stets nur eckenverknüpft vorkommen.
Bei der in-vitro-Messung von zum Beispiel der Aktivität einer Silaffin-Fraktion sollte im Idealfall von ortho-Kieselsäure ausgegangen werden, die jedoch als Reinstoff nicht verfügbar ist. Die übliche Technik besteht in der Verwendung hydrolyseinstabiler Kieselsäureester als Si-Quelle. Am häufigsten werden Tetramethoxysilan (TMOS) und Tetraethoxysilan (TEOS) verwendet. Dabei tritt jedoch eine Schwierigkeit auf, die auf der anderen Seite zu interessanten Schlussfolgerungen führt. Die Hydrolyseempfindlichkeit ist nämlich nicht sehr stark ausgeprägt [silica4], so dass die Reaktivität von zum Beispiel SiCl4 bei Weitem nicht erreicht wird. Perry kritisiert in [silica2] die sich über Minuten und Stunden hinziehende Kieselsäureester-Hydrolyse und verwendet für ihre eigene Studie zur Aminosäure/Kieselsäure-Wechselwirkung ein Tris(catecholato)silicat/Säure-Gemisch, um ohne Verzögerung ortho-Kieselsäure in einer wässrigen Lösung bereitzustellen.
Die Ursache der langsamen und schrittweisen Hydrolyse für die Suche nach bislang unerkannten „Cofaktoren“ aufschlussreich: die Reaktion ist keineswegs kinetisch besonders gehemmt, es fehlt offensichtlich vielmehr die thermodynamische Triebkraft. So konnte Kinrade zeigen, dass in einer alkalischen Wasser/Methanol-Mischung Silicat teilweise zu Methylsilicat reagiert. Si-O-C-Verknüpfungen sind also keineswegs so hydrolyseempfindlich wie dies lange angenommen worden ist, so dass die Suche nach Bioliganden für die Siliciumkomplexierung nicht aussichtslos erscheint.
Im Tierreich ist Verkieselung bei den Schwämmen von besonderer Bedeutung. Schwämme bilden SiO2-Skelettbestandteile aus, die wesentlich härter, dichter und schwerer löslich sind als Kieselalgenschalen. Neben den auffälligen Skelettnadeln (engl. spicules) treten kunstvoll geformte SiO2-„Klammern“ auf, die beim Aufbau des Skeletts als Verbinder dienen. [silica3] zeigt die Formenvielfalt der von Schwämmen hergestellten Bauelemente, deren Funktion nicht allein der Aufbau eines stützenden Skelettes ist.
Die Silicifizierung geht von einem axialen Filament aus, einem Protein, das die Verkieselung steuert. Die Kieselsäureablagerung beginnt mit dem Einfüllen nanometergroßer Kieselsäurekugeln („Nanokugeln“), die dabei zu größeren Kugeln zusammentreten. Fig. 21 in [silica3] zeigt zwei Skelettbausteine in verschiedenem Verkieselungszustand. Die linke der beiden „Klammern“ hat eine erkennbar partikuläre Struktur, die bei der rechten, älteren Klammer durch amorphe Kieselsäure verkittet ist und dadurch glatt wirkt. Die Funktion solcher Verbindungsklammern (Mikroskleren) besteht in der Verbindung der Skelettnadeln (Makroskleren). Dem sich daraus ergebenden flexiblen Skelettaufbau (vgl. Fig 29F in [silica3]) steht in anderen Schwämmen ein festgefügtes SiO2-Skelett gegenüber (Fig 4 in [silica3]).
Das Protein des axialen Filaments in Schwamm-Skelettnadeln lässt sich durch Flusssäurebehandlung vom SiO2 befreien. Es werden zwei Hauptkomponenten gefunden, α- und β-Silicatein. Die Sequenzanalyse zeigt die Verwandtschaft der Silicateine mit den Enzymen der Cathepsin-Familie – Proteasen, die in den Osteoclasten für den Kollagenabbau zuständig sind. Es liegt keine Strukturanalyse vor. Aus Modellierungsstudien wird geschlossen, dass vor allem Serin-Seitenketten, unterstützt von Histidinresten als Base, ein Siliciumzentrum nukleophil angreifen können und so die Polykondensation (und auch die Hydrolyse von Kieselsäurestern) katalysieren. Eine in-vitro-Aktivität von Aminosäuren auf die Kieselsäure-Polykondensation ist dabei nicht überzeugend nachzuweisen [silica2].
Auch die biochemische Seite der Silicateinwirkung ist kaum verstanden. Es scheint ein Zusammenspiel mit Ferritin vorzuliegen, das als innerer Kern des axialen Filament gesehn wird (Fig. 9 in [silica5]).
Isoliertes Silicatein katalysiert die Kieselsäure-Polykondensation, die typischen Aminosäuren des Silicateins und kurzkettige Lysin-Oligomere tun dies nicht. Diese Ergebnisse führen derzeit zu zwei verschiedenen gedanklichen Ansätzen, zum Beispiel SiO2-Kugeln nach dem Vorbild der Skelettnadel-Baueinheiten zu synthetisieren. Der erste Weg nutzt das Protein selbst. So wurde Silicatein mit dem Ziel auf Oberflächen immobilisiert, diese mit strukturiertem SiO2 belegen (Fig. 3 b und c in [silica6]). Die Versuche reichen über SiO2 hinaus. Ein dem Silicatein ähnliches Protein wurde in Pilzen gefunden (die nicht verkieseln!), und für die Synthese von ZrO2-Nanopartikeln genutzt (Fig. 1 in [silica7]).
Eine zweite Idee betrifft die katalytische Wirkung funktioneller Gruppen des Silicateins oder der Silaffine. In [silica8] wird über die Katalyse der Kieselsäure-Polykondensation berichtet, wenn zahlreiche kleinmolekulare, bifunktionelle Verbindungen untersucht werden. Als wirksamer Katalysator stellt sich dabei Cysteamin heraus (Figs. 1 und 4 in [silica8]), bei dem man die Seitenketten von Lysin und Cystein kombinert sehen kann – wenn man denn alles aus dem biochemischen Blickwinkel sehen will.
Was sind konkret die Ziele bei den Versuchen, sphärische SiO2-Partikel herzustellen. Ein besonders interessantes Ziel ist die biomimetische Synthese neuer Opal-Varianten. Opale, die eine kubisch-flächenzentrierte Anordnung sub-μm-großer hydratisierter SiO2-Partikel darstellen, haben eine Fülle von Anwendungen [silica9], siehe dort Fig. 1 für eine Abbildung des Opal-Aufbaus. Eine besonders interessante Verwendung ist die Nutzung als photonische Kristalle [silica11]. Für deren Aufbau ist außer den SiO2-Kugeln das umgekehrte Prinzip genutzt worden, nämlich der Synthese eines inversen Opals, dessen Konstruktion eine weitere Anwendung der Kieselsäure-Polykondensation ist (Fig. 6 in [silica10], [silica11].
Üblicherweise wird ortho-Kieselsäure als die Transportform des Siliciums angesehen. Es ist daher bemerkenswert, das Schwämme über ein Silicase genanntes Enzym verfügen. Auch für dieses sind keine Strukturdaten bekannt, es wird jedoch als der Carboanhydrase-Familie zugehörig beschrieben.
M. Sumper, N. Kröger: Silica formation in diatoms: the function of long-chain polyamines and silaffins: J. Mater. Chem. 2004, 14, 2059–2065 [silica1].
D. Belton, G. Paine, S. V. Patwardhan, C. C. Perry: Towards an understanding of (bio)silicification: the role of amino acids and lysine oligomers in silicification. J . Mater. Chem. 2004, 14, 2231–2241 [silica2].
M.-J. Uriz, X. Turon, M. A. Becerro, G. Agell: Siliceous Spicules and Skeleton Frameworks in Sponges: Origin, Diversity, Ultrastructural Patterns, and Biological Functions. Micr. Res. Techn. 2003, 62, 279–299 [silica3].
F. Brunet, B. Cabane, M. Dubois, B. Perly: Sol-Gel Polymerization Studied through 29Si NMR with Polarization Transfer. J. Phys. Chem. 1991, 95, 945–951 [silica4].
W. E. G. Müller, A. Krasko, G. Le Pennec, H. C. Schröder: Biochemistry and Cell Biology of Silica Formation in Sponges. Micr. Res. Tech. 2003, 62, 368–377 [silica5].
M. N. Tahir, P. Théato, W. E. G. Müller, H. C. Schröder, A. Janshoff, J. Zhang, J. Huthe, W. Tremel: Monitoring the formation of biosilica catalysed by histidine-tagged silicatein. Chem. Comm. 2004, 2848–2849 [silica6].
V. Bansal, D. Rautaray, A. Ahmad , M. Sastry: Biosynthesis of zirconia nanoparticles using the fungus Fusarium oxysporum. J. Mater. Chem. 2004, 14, 3303–3305 [silica7].
K. M. Roth, Y. Zhou, W. Yang, D. E. Morse: Bifunctional Small Molecules Are Biomimetic Catalysts for Silica Synthesis at Neutral pH. J. Am. Chem. Soc. 2005, in press [silica8].
M. R. Newton, A. K. Bohaty, H. S. White, I. Zharov: Chemically Modified Opals as Thin Permselective Nanoporous Membranes. J. Am. Chem. Soc. 2005, 127, 7268–7269 [silica9] .
Z. Zhou, X. S. Zhao: Opal and Inverse Opal Fabricated with a Flow-Controlled Vertical Deposition Method. Langmuir 2005, 21, 4717–4723 [silica10].
J. Ozin: The photonic opal – the jewel in the crown of optical information processing. Chem. Comm. 2003, 2639–2643 [silica11].
[4Fe-4S]-Ferredoxin-Zentren durch Selbstorganisation? Modelle: ja, mitochondriale Ferredoxine werden jedoch durch Metallchaperone unter strikter Kontrolle biosynthetisiert.
Idee: die ersten „biochemischen“ Reaktionen fanden an Eisensulfiden statt.
Aktuelle Ergebnisse: [fes3] zeigt die Bildung von NH3 aus N2 und H2S, treibende Kraft ist die Pyrit-Bildung.
[fe4] zeigt die Bildung carbonylierter Produkte aus einfachen Edukten wie Alkylthiolen und CO unter FeS-Katalyse.
Zu den von Eisensulfid ausgehenden Versuchen erregen derzeit Strukturanalsen Aufsehen, welche die Palette reaktiver Metall-Schwefel-Cluster über die Ferredoxine und über die Nitrogenase-Cluster hinaus erweitern.
Ein Beispiel ist Carboxidothermus-hydrogenoformans-CO-Dehydrogenase (PDB-Code 1SU6).
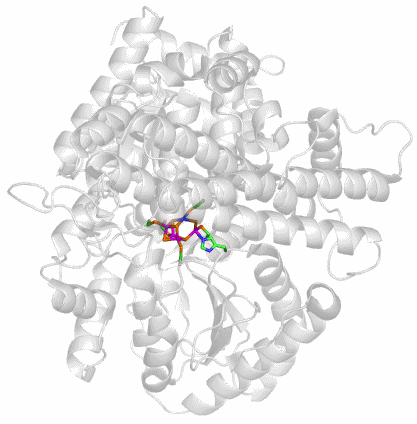
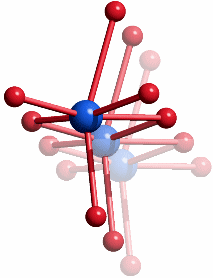
Das aktive Zentrum ist ein ungewöhnlicher [4Fe-Ni-5S]-Cluster, der über vier Cys und ein Fe-gebundenes His mit dem Protein verbunden ist:
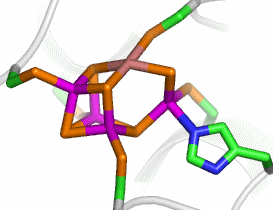
Der Cluster enthält ein quadratisch-planar koordiniertes Nickelatom. Der Mechanismus der CO2-Reduktion ist unbekannt.
Verwandte Zentren sind in [fes2] zusammengestellt, darunter der Acetyl-CoA-erzeugende A-Cluster von CODH/ACS und die Nitrogenase-Cluster.
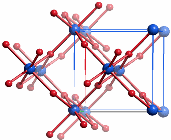
Solche Strukturen laden zum Vergleich mit Fragmenten von Metallsulfidstrukturen ein, zum Beispiel mit nickelhaltigem Greigit:
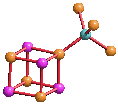
[fes1] erörtert, dass die notwendigen Reaktionsbedingungen in geologischer Umgebung vorliegen.
M. J. Russell, W. Martin: The rocky roots of the acetyl-CoA pathway. Trends Biochem. Sci. 2004, 29, 358–363 [fes1].
C. L. Drennan, J. W. Peters: Surprising cofactors in metalloenzymes. Current Opinion in Structural Biology 2003, 13, 220–226 [fes2].
M. Dörr, J. Käßbohrer, R. Grunert, G. Kreisel, W. A. Brand, R. A. Werner, H. Geilmann, C. Apfel, C. Robl, W. Weigand: Eine mögliche präbiotische Bildung von Ammoniak aus molekularem Stickstoff auf Eisensulfidoberflächen. Angew. Chem. 2003, 115, 1579–1581 [fes3].
G. D. Cody, N. Z. Boctor, T. R. Filley, R. M. Hazen, J. H. Scott, A. Sharma, H. S. Yoder Jr.: Primordial Carbonylated Iron-Sulfur Compounds and the Synthesis of Pyruvate. Science 2000, 289, 1337–1340 [fes4].
E. Bäuerlein (Ed.): Biomineralization: Progress in Biology, Molecular Biology and Application. 2nd Edition. Wiley-VCH 2004
Zusammenfassung hieraus zu Magnetit, Calciumcarbonat und Siliciumdioxid: E. Bäuerlein: Biomineralisation von Einzellern: eine außergewöhnliche Membranbiochemie zur Produktion anorganischer Nano- und Mikrostrukturen. Angew. Chem. 2003, 115, 636–664 [biomin1].
M. Epple: Biomineralien und Biomineralisation. Teubner Studienbücher Chemie 2003
S. Mann: Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry. Oxford Chemistry Masters 2001