 Biomineralisation
Biomineralisation 2 × 2 Stunden im Rahmen der interdiszilinären Ringvorlesung „Biomineralisation“, Di 16–18, HS3, Luisenstraße 37
Beiträge im SS 2006:
16. Mai 2006: Bioanorganische Grundlagen der Biomineralisation: Ferritin
11. Juli 2006: Biogenes Siliciumdioxid
Bitte beachten Sie die Hinweise auf die Protein Data Bank (PDB) im Skript der Vorlesung Bioanorganische Chemie.
Kristalldaten, bei denen keine Quelle angegeben ist, stammen aus der ICSD (Inorganic Crystal Structure Database).
Informationen zu Raumgruppen finden sich unter der angegebenen Nummer in den International Tables for Crystallography.
Mit einer Menge von 3–5 g ist Eisen im menschlichen Körper das wichtigste Übergangsmetall, an das von der Menge her nur noch Zink (ca. 2 g) in etwa heranreicht. Die Hauptmenge von ca. 2/3 ist im Hämoglobin gebunden. Das nächstgrößere Einzelvorkommen ist das Speichereisen, Ferritin, in dem der Körper das Metall für die zahlreichen Funktionen der unterschiedlichsten Eisenzentren vorhält. Der Oxoeisenkern des Ferritins ist neben dem biologischen Apatit der Knochen und Zähne und dem Calciumcarbonat der Otolithen das dritte Biomineral unseres Körpers. In der Literatur wird eine enge Verwandschaft zwischen dem Ferritinkern, der mehr als 4000 Eisenatome umfassen kann, und dem wenig kristallinen Mineral Ferrihydrit gesehen. Der Weg des Eisen von der Nahrung bis in den Ferritinkern ist typisch für ein schwerlösliches Mineral, das überdies einen toxischen Baustein enthält: Mobilisierung durch eine spezialisierte Transportform, strikte Kontrolle der Zustandsform im Körper, Einstellen der notwendigen Oxidationsstufe für die Mineralisation, und schließlich die templatinduzierte Biomineralisation selbst.
Eisen ist wie Mangan, Cobalt, Nickel und Kupfer ein Übergangsmetall. Es kommt in den Oxidationsstufen +ii und +iii in der Natur vor. Durch das Auftreten in zwei benachbarten Oxidationsstufen wird Eisen in Organismen neben dem Sauerstofftransport für die Katalyse von 1-Elektronen-Redoxvorgängen verwendet (Ferredoxine, Cytochrome). In Abwesenheit spezieller Liganden und in Anwesenheit von Sauerstoff ist die Eisen(iii)-Form die stabilere. Eisen(iii) kommt im Gegensatz zu Calcium, Magnesium oder Zink bei physiologischem pH-Wert nicht einfach in hydratisierter Form vor. Hierzu ist das [Fe(H2O)6]3+-Ion viel zu sauer. Durch die Abspaltung von Protonen liegen um pH 7 herum stattdessen Hydroxide mit wechselndem Wassergehalt vor, „Fe(OH)3“. Die oft verwickelte Verknüpfung von Säure-Base-Gleichgewichten mit der Chemie von Oxidations- und Reduktionsvorgängen lässt sich am anschaulichsten mit graphischen Hilfsmitteln wie zum Beispiel den Pourbaix-Diagrammen darstellen.
Elektrochemische Potentiale, bei deren Formulierung Komplexverbindungen auftreten, sind pH-abhängig, wenn die Liganden protonierbar oder deprotonierbar sind oder wenn Liganden bei Änderung des pH-Wertes substituiert werden. Die Aqua-Liganden hydratisierter Metall-Ionen sind deprotonierbar. Die Folge ist eine elektrostatische Stabilisierung der höheren Oxidationsstufe, das elektrochemische Potential sinkt mit dem pH-Wert, die oxidierende Wirkung der höheren Oxidationsstufe nimmt ab. Quantitativ wird dies durch die Nernstsche Gleichung beschrieben. Diese dient, zusammen mit den Löslichkeitsprodukten eventuell auftretender schwerlöslicher Phasen, zur Konstruktion von Potenital-pH-Diagrammen, den sogenannten Pourbaix-Diagrammen.
Zahlenangaben zur Konstruktion des folgenden Diagramms: E0(Fe2+/3+) = 0.77 V; c(Fe) = 10−6 m. Löslichkeitsprodukte: Fe(OH)3 10−39; Fe(OH)2 10−16:
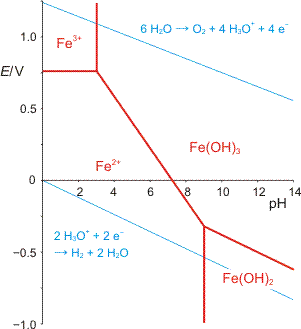
Es wird deutlich, dass Eisen trotz seiner Häufigkeit alles andere als ein für Organismen leicht verfügbares Spurenelement ist. Auf die Frage, warum im Verlauf der Evolution Eisen trotz dieser gravierenden Einschränkung seine zentrale Bedeutung erlangen konnte, wird im letzten Abschnitt eingegangen.
Die Literatur enthält widersprüchliche Meinungen zur Bedeutung aktiver Eisenaufnahmewege in Bakterien. Man bedenke die oft reduzierenden Bedingungen in den Lebensräumen dieser Organismen, mit denen die Verfügbarkeit von Eisen(ii) einhergeht. Zur Sicherstellung des Eisenbedarfs scheiden die meisten Bakterien jedoch Stoffe aus, mit denen sie dreiwertiges Eisen mobilisieren können. Generell aquirieren Bakterien in aggresiver Weise das von ihnen benötigte Eisen aus ihrer Umgebung. Die hierzu genutzten zahlreichen (ca. 200 bekannte) „Siderophore“ enthalten Liganden mit hoher Eisen(iii)-Affinität. Meist handelt es sich um Catecholate und Hydroxamate, die in deprotonierter Form extrem stabile Eisen(iii)-Komplexe bilden. Die Siderophore humanpathogener Keime müssen mit dem menschlichen Eisen-Transportprotein Transferrin konkurrieren. Transferrin bildet einen so stabilen Eisen(iii)-Komplex, dass sich die Plasmakonzentration an freiem Eisen(iii) auf ca. 10−24 m berechnet – was einer einstelligen Zahl an freien Eisen(iii)-Ionen im gesamten Blutplasma entspricht.
Das am besten untersuchte Siderophor ist Enterobactin (Ent), das drei Catecholato(2−)-Liganden zum Aufbau einer FeO6-Umgebung zur Verfügung stellt. FeEnt wird durch FepA, einer Proteinpore in der äußeren Bakterienmembran erkannt und in die Zelle transportiert. Die Abbildung von FepA basiert auf dem PDB-Eintrag 1FEP. Der aus aus ca. 700 Aminosäuren bestehende, lange Proteinstrang bildet nicht nur die Wand der Pore, sondern im Inneren ist ein Verschluss sichtbar, der sich nur für FeEnt öffnet.
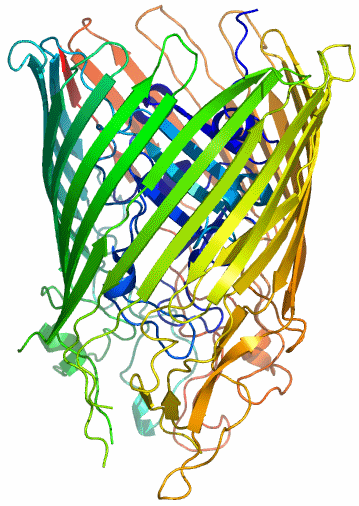
Wie erkennt FepA, ob Ent im Inneren ein Eisen-Zentralatom trägt oder ob es leer ist? Die Information hierzu beruht vor allem auf Modellierungsstudien. Bis heute ist FeEnt nicht kristallisiert worden, 1992 wurde jedoch die Struktur des Ent-Vanadium(iv)-Komplexes publiziert, so dass eine Grundlage für die Modellierung vorhanden ist. Fig. 1 in [magnetite1] zeigt eine Abbildung von beladenem und von unbeladenem Ent. In Abwesenheit von Eisen sind die drei auf einem Cyclotriserin-Ring fixierten Brenzcatechin(„Catechol“)-Einheiten nach außen gerichtet. Wasserstoffbrückenbindungen des Typs O(Catechol)–H···O(Amid) halten die eisenchelatisierende Funktion in dieser Stellung. Die Bindung eines Eisen(iii)-Ions führt zur Deprotonierung des Catechols, worauf der Ligand zusammen mit dem Eisen-Ion eine 180°-Drehung in das Siderophor-Innere ausführt und durch eine N–H···O(Catecholat)-Bindung „einrastet“. Das aktive „Einsammeln“ von Eisen-Ionen verlangt den nach außen weisenden potentiellen Chelat-Ligand. Die bei Metalloproteinen übliche Gestaltung einer Metallbindungsstelle aus einzelnen Aminosäure-Seitenketten würde dies nicht leisten können.
Nachdem FeEnt in die Zelle eingeschleust ist, wird das Eisen-Ion durch Zerstörung des Transportmoleküls freigesetzt. Hierzu wird die Amidbindung hydrolysiert.
Der überragenden Bedeutung des Eisens als Bioelement steht eine äußerst geringe Bioverfügbarkeit gegenüber. Im Verlauf der Evolution reicherte sich die Erdatmospäre mit Sauerstoff an, durch den das an der Boden- oder Gewässeroberfläche vorliegende Eisen in die Oxidationsstufe +iii gebracht wurde – aus dem präbiotischen Eisen(ii)-sulfid wurde Eisen(iii)-(hydr)oxid. Aus dem Löslichkeitsprodukt von maximal ca. 10−38 für frisch gefälltes „Eisen(iii)-hydroxid“ errechnet sich bei pH 7 eine Eisen(iii)-Konzentration von maximal 10−17 mol L−1.
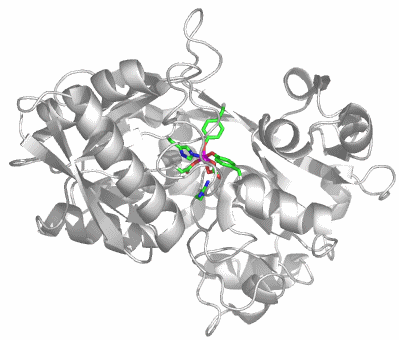
Transferrine binden Eisen(iii) mit hoher Bindungskonstante. Das Eisen-Transportprotein im menschlichen Blut ist das Serum-Transferrin (sTf). Die Strukturanalyse zeigt, dass die Natur für den Transport von zwei Eisen(iii)-Atoms ein großes, einsträngiges Protein mit fast 700 Aminosäuren aufbaut, das aus zwei ähnlichen Hälften besteht. Das Bild zeigt die Strukur der N-terminalen Hälfte von humanem sTf (PDB-Code: 1D3K):
Das aktive Zentrum zeigt die Ursache der CO2-Abhängigkeit des Transferrins:
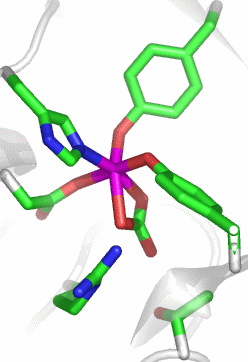
Das FeIII(CO3)-Fragment ist perfekt in das Apoprotein eingebunden. Zwei der sechs Koordinationsstellen des Eisen(iii)-Zentralatoms sind durch einen Carbonato-Liganden belegt. Die hohe Stabilität des Komplexes scheint hiermit nicht im Einklang zu sein, man beachte jedoch die Einbindung des Carbonats in die Gesamtstruktur durch Wasserstoffbrückenbindungen und den weitgehenden Ladungsausgleich. Zwei der Wasserstoffbrückenbindungs-Donoren sind im Bild gezeigt: links unter dem Carbonat-Ion eine kationische Arginin-Seitenkette, die bei physiologischem pH-Wert protoniert vorliegt, außerdem rechts unten die Hydroxylfunktion einer Threonin-Seitenkette. Weitere H-Brücken gehen von Amidfunktionen der Proteinhauptkette aus. Zwei der übrigen Liganden gehören zu den häufigsten metallbindenden Aminosäureseitenketten: Aspartat und Histidin. Die verbleibenden zwei Koordinationsstellen sind demgegenüber durch einen weniger häufigen Liganden besetzt, nämlich durch Tyrosinat.
Wie bei Enterobactin ist die Eisenaufnahme auch bei Transferrin ein Schaltvorgang. Transferrin steht in Konkurrenz zu Siderophoren. Wie Enterobactin ist auch bei Transferrin die Eisenbindung oder -freisetzung durch eine Konformationsänderung optimiert. Bei der Freisetzung öffnet sich der im Bild des Holoenzyms sichtbare Graben oberhalb der Eisenbindungsstelle. Umgekehrt „beißt“ das geöffnete Enzym bei der Bindung eines Eisenatoms an die zuvor mit Carbonat beladene Enzymtasche zu. Die molekulare Ursache des Schaltprozesses ist Gegenstand der Diskussion. Bedeutung wird einem pH-Abfall bei der Bindung an den Transferrin-Rezeptor beigemessen, der die mehrfache Protonierung der Eisenbindungsstelle bewirkt, beginnend mit einer Carbonat-Protonierung. Das Prinzip des Zugreifens zweier beweglicher Proteinteile als Folge eines Schaltsignals wird in [ferritin3] näher beleuchtet.
Transferrine übertragen das herantransportierte Eisen an Ferritine, in denen Oxoeisen(iii)-Kerne ein ca. 4000 Eisenatome umfassendes Biomineral bilden (Fig. 2 in [ferritin 1]).
In den Apo-Ferritinen umschließen Untereinheiten große Hohlräume [ferritin4,6]. Alle Ferritine sind in symmetrischer Weise aus einem strukturell kaum variierten Grundbaustein aufgebaut. Es handelt sich um eine ca. 170 Aminosäuren lange Proteinkette, in der vier α-Helices nahezu parallel zueinander ausgerichtet sind. Bakterien- und Pflanzenferritine sind aus 12 oder 24 gleichartigen Proteinsträngen zusammengesetzt. Im O-symmtrischen (Punktgruppe 432 nach Hermann-Mauguin) 24-mer wird so ein Hohlraum umschlossen, der ca. 8 nm Durchmesser hat (Fig. 1 in [ferritin 1]). Es existieren keine Strukturanalysen, die sowohl das Apoprotein als auch einen wohlstrukturierten Eisenkern zeigen. Die derzeit publizierten ca. 40 Ferritinstrukturen sind vielmehr im wesentlichen Apoproteinstrukturen, wobei einzelne Metallbindungsstellen sichtbar werden, wenn die intakten Kristalle das Eintauchen in verschiedene metallsalzhaltige Lösungen tolerierten und dabei einige Metallionen eingelagert haben. Tierferritine bestehen oft aus zwei verschiedenen Proteinsträngen. So sind die 24-mere von menschlichem Ferritin aus H- oder L-Ketten aufgebaut, die Ferroxidase-Zentren aufweisen (H) oder nicht (L). Ein auffallendes Merkmal ist die oft geringe Sequenzhomologie der Ferritine: obwohl die Vier-Helix-Gestalt ubiquitär ist, unterscheiden sich die Aminosäuresequenzen oft erheblich voneinander. Bakterioferritine sind ebenfalls 24-mer, wobei diese Hämeisen-Zentren tragen können oder nicht. Neben diesen „großen“ Ferritinen werden bei Bakterien kleine, dodekamere Ferritine gefunden, welche die Dps-Unterfamilie bilden und deren Kern ca. 500 Eisenatome im ca. 4.5 nm großen Hohlraum enthalten. „Dps“ steht für „DNA-binding protein from starved cells“. Dieser Ferritintyp wird von Bakterien unter Hungerbedingungen oder unter oxidativem Stress produziert, wobei dichte Schichten von Dps-Partikeln im Wechsel mit eingeschlossenen und dadurch geschützten DNA-Schichten gefunden werden. Der Schutz für die DNA ist nicht unspezifisch zum Beispiel im Sinne einer Diffusionsbarriere zu verstehen. Dps oxidiert vielmehr das in Gegenwart von Sauerstoff oder H2O2 toxische Eisen(ii) unter H2O2-Verbrauch zu eingelagertem Eisen(iii). Man vergleiche mit den Ferroxidase-Zentren von 24-mer Ferritinen, die Sauerstoff für die Eisen(ii)-Oxidation verbrauchen und H2O2 freisetzen.
Stichworte: Eingangstrichter für hydratisierte Eisen(ii)-Ionen (Figure 3), Ferroxidase-Zentren innerhalb von α4-Stapeln (Figure 4[f]), Metallatom-Pfad vom Ferroxidase-Zentrum in den inneren Hohlraum (Figure 5).
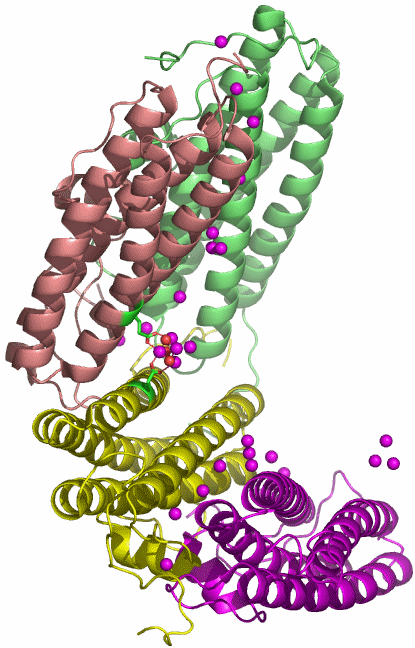
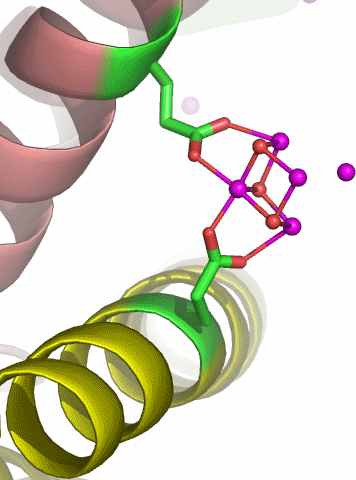
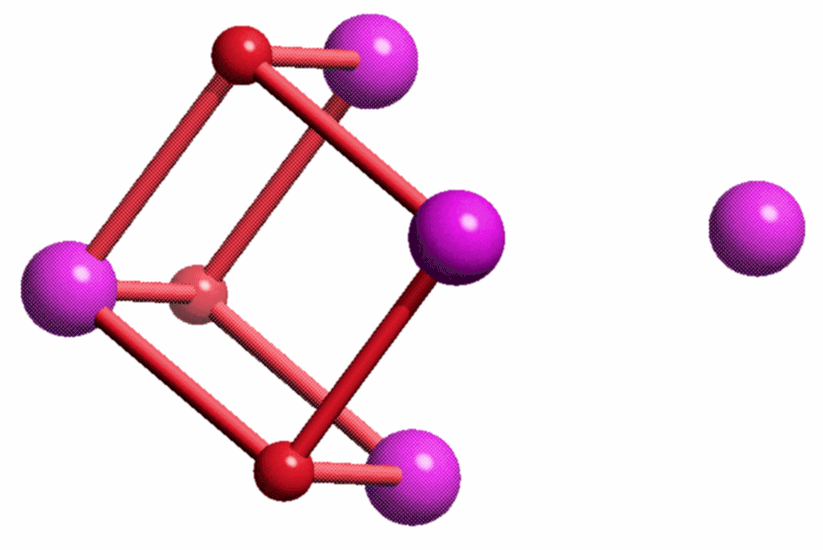
Der PDB-Eintrag 1TKP zeigt die asymmetrische Einheit trigonaler Kristalle von Halobacterium-salinarum-Ferritin in einer Auflösung von 2.2 Å. H. salinarium ist ein halophiles Archäon. Die asymmetrische Einheit des dodekameren Proteins enthält vier Einzelstränge, die in verschiedener Farbe dargestellt sind. Das Tetramer mit den deutlich erkennbaren vier α-Helices eines jeden Einzelstranges ist gebogen und schließt mit zwei weiteren Tetrameren einen T-symmetrischen Hohlraum ein (Punktgruppe 23 nach Hermann-Mauguin, die Symmetrie ist bei 1TKP nicht-kristallographisch). Unter den publizierten Ferritinstrukturen ist 1TKP diejenige, die den Templatcharakter des Proteins am deutlichsten zeigt. Nach Eintauchen der Kristalle in eine Eisen(ii)-Lösung unter Luftzutritt gelingt es, den Hohlraum des Dodekamers mit 110 Eisenatomen zu füllen. Die Fe-Atome der asymmetrischen Einheit sind im Bild violett dargestellt; sie finden sich auf der konvexen Seite. Besonders instruktiv ist die in ferritin1 als Nukleationszentrum NII bezeichnete Position, die im unteren Bild vergrößert dargestellt ist. Ungefähr waagrecht verläuft eine nichtkristallographische C3-Achse, welche die beiden dargestellten Helices um eine dritte ergänzt, die vor dem Oxoeisen-Fragment liegt und deshalb der Übersicht halber weggelassen wurde. Beachtet man also, dass die beiden dargestellten Glutamat-Reste um einen dritten ergänzt werden, so ergibt sich ein C3-symmetrisches Templat mit insgesamt 6 Haltepunkten. An dieses Templat binden 4 Eisenatome in Form eines Tetraeders derart, dass die drei den Glu-Resten zugewandten Tetraederkanten im geläufigen μ-O1,O2-Bindungsmodus koordinieren. Werden die einzelnen O-Atome im Bild als Hydroxo-Liganden interpretiert, so ergibt sich insgesamt ein Fe4(μ3-OH)3-Motiv. Die Strukturanalyse zeigt nicht, wie das vierte Eisenatom mit dem Rest des Oxoclusters verbunden ist.
Das sich ergebende Muster lässt sich als Ausschnitt aus einer Festkörperstruktur betrachten, wenn Eisen- und Sauerstoff-Atome im Sinne einer anti-NiAs-Struktur auf Arsen- und Nickelplätze gelegt werden, Eisenatome also dicht gepackt werden und Sauerstoffatome in Oktaederlücken gelegt werden:
Strichworte: Der Ferritinkern gesunder Menschen besteht im wesentlichen aus Ferrihydrit und anderen Eisen(iii)-oxid-Phasen, darunter auch Hämatit. Neurodegenerative Erkrankungen wie die Alzheimersche Krankheit oder die progressive supranukleäre (Blick-)Lähmung gehen mit einer verminderten Ferrihydrit-Ablagerung im Hirn-Ferritin einher, zugleich werden vermehrt Eisen(ii,iii)-oxide gefunden (Magnetit und Wüstit, Fe1−xO, x ca. 0.9)
Hämatit kristallisiert in der Korundstruktur. Der charakteristische Baustein sind kantenverknüpfte FeO6-Oktaeder, die ein hexagonales Wabennetz aufbauen:
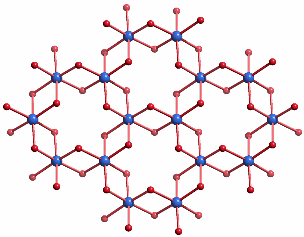
Goethit, α-FeOOH:
P b n m (Nichtstandardaufstellung der Raumgruppe Nr. 62 P n m a; P b n m entspricht der Achsenwahl c a b)
a = 4.605, b = 9.960, c = 3.023 Å, alle Atome in 4 c x, y, ¼ mit den folgenden x- und y-Werten:
Fe 0.04770 0.85390
O1 0.70800 0.19960
O2 0.20400 0.05310
H 0.4095 0.0876
Projektion entlang ca. [001]:
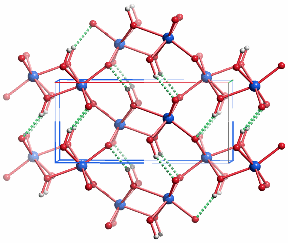
In der Goethit-Struktur sind alle Eisen(iii)-Zentren verzerrt oktaedrisch koordiniert. Entlang [001] sind die FeO6-Oktaeder kantenverknüpft, ferner tritt zwischen jeweils zwei Oktaedersträngen paarweise Kantenverknüpfung auf. Die Struktur lässt sich von der Rutil-Struktur durch „kristallographische Scherung“ ableiten, durch welche die Voraussetzung für die Ausbildung von Wasserstoffbrückenbindungen gegeben wird (im bild grün gestrichelt).
Ferrihydrit ist ein wenig kristallines Eisen(iii)-(hydr)oxid, dessen wesentliches Bauelement in einer doppelt-hexagonalen Packung von Sauerstoffatomen (Schichtfolge ABAC…) mit Eisen in einem Teil der Oktaederlücken angesehen wird. Die Zusammensetzung wird oft mit 5 Fe2O3 · 9 H2O angegeben, entsprechend Fe5O3(OH)9. Auf 12 packungsbildende (Hydr)oxid-Ionen kommen also 5 Eisenatome; die Oktaederlücken sind also ungefähr zur Hälfte (5/12) besetzt [ferritin5]. Das in der eisenreichen Dsp-Struktur gefundene Baumotiv kommmt also in der derzeit akzeptierten Ferrihydrit-Struktur nicht vor.
Akaganéit, β-FeOOH, ist ein in den letzten Jahren intensiv untersuchtes Mineral der Hollandit-Familie, A2B8(O,OH)16, A = Ba, K, Pb; B = Fe, Mn; die A- und B-Lage ist oft unterbesetzt. Hollandit ist tetragonal und leitet sich von der Rutilstruktur ab. Die angegebene Formel FeOOH ist idealisiert. Eine realere Formulierung ist zum Beispiel FeO5/6(OH)7/6Cl1/6. Akaganéit wird vor allem dann gefunden, wenn große Ionen wie Cl−, CO32− oder MoO42− im Sinne einer anti-Hollandit-Struktur Kanäle entlang [001]tetragonal besetzen. Möglicherweise kann auch OH− diesen Platz einnehmen. Aktuelle Studien an Akaganéit verfeinern die Struktur nur mit monokliner Symmetrie [ferritin8,ferritin9]. Das Bild zeigt das Ergebnis der in [ferritin8] beschriebenen Neutronbeugungsuntersuchung in einer Projektion ungefähr entlang [010]. Die grünen Kreise zeigen die Lagen von Chlorid-ionen an, deren Besetzungsfaktor kleiner als ½ ist. Zum Ladungsausgleich liegt irgendwo in der Struktur die äquivalente Menge an Protonen vor, deren Lage aber auch durch Neutronenbeugung nicht zufriedenstellen bestimmt werden konnte. Werden die nicht zugeordneten Protonen und die Chlorid-Ionen weggelassen, so ist die Zusammensetzung der übrigen Kristallbausteine wie bei Goethit FeOOH.
Das Verknüpfungsmotiv der FeO6-Polyeder ist in den beiden FeOOH-Modifikationen ähnlich. Auch in Akaganéit sind verzerrte FeO6-Oktaeder über Kanten zu Strängen verknüpft. Jeweils zwei Stränge sind durch erneute Kantenverknüpfung miteinander verbunden. Der Unterschied liegt bei der Funktion der Wasserstoffatome. Im Goethit bilden diese Brückenbindungen zu Oxid-Ionen aus, während sie beim Akaganéit in die entlang [010] verlaufenden, aniongefüllten Kanäle hineingerichtet sind.
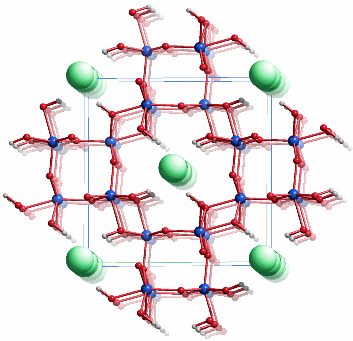
Ein stärker gegen [010] geneigter Ausschnitt hebt die Kantenverknüpfung der FeO6-Oktaeder entlang [010] hervor:
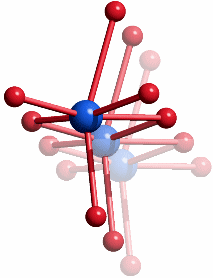
Das Motiv von zu Strängen kantenverknüpfter Oktaeder ist in der prototypischen Struktur des Rutils vorgegeben. Die Abbildung zeigt einen entlang [001] projizierten Ausschnitt der tetragonalen AB2-Struktur, der die Verwandtschaft zum Akaganéit besonders hervorhebt:
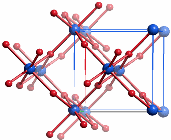
Beide Prototypen, Korund und Rutil sind durch kantenverknüpfte Oktaeder geprägt. Dieses Baumotiv wird auch in mehrkernigen Komplexen dreiwertiger Metalle mit Oxo-Liganden oft gefunden. Die Vermutung ist vernünftig, die oktaedrische Umgebung der Metall-Ionen sowie die Verknüpfung über Ecken und Kanten, nicht jedoch über Flächen, auch in den amorphen Phasen als strukturbestimmend anzunehmen.
Amorphe, ligandstabilisierte Oxoeisen(iii)-Phasen, deren Struktur den oben zusammengestellten Bauprinzipien folgen sollte, werden seit langem in der Medizin eingesetzt. Sie entstehen beim Neutralisieren saurer Eisen(iii)-Salzlösungen auf pH-Werte zwischen 7 bis ca. 9 anstelle brauner Niederschläge, wenn den Lösungen vor dem Fällen Kohlenhydrate zugegeben wurde. Unter diesen Bedingungen entstehen keine Niederschläge, sondern kaffeebraune Lösungen, die zur Trockne eingeengt werden können und anschließend bei Wasserzugabe wieder dieselben Lösungen ergeben. Lichtstreuversuche ergeben Partikelgrößen von ca. 2 nm. Das Bild zeigt eine solche Lösung, in der Eisen(iii) durch weniger als 20 Mol-% d-Mannit an der Ausfällung gehindert wird:

Bereits in den 40er Jahren des vergangenen Jahrhunderts tauchten Präparationen in den Arzneibüchern auf, die Rohrzucker (Saccharose, engl. sucrose) zu diesem Zweck nutzten. Sie wurden zur Therapie des Eisenmangels oral verwendet. Die Verfügbarkeit des Eisen bei dieser Art der Anwendung ist offensichtlich sehr begrenzt. Eine Renaissance erleben solche Zubereitungen seit der Verfügbarkeit von Erythropoietin (EPO), das vor allem bei der Bekämpfung der Blutarmut von Dialysepatienten eingesetzt wird. Die Therapie mit EPO verlangt hohe Eisengaben, die durch Aufnahme aus der Nahrung nicht zuverlässig zu decken sind. Die nur begrenzte Verfügbarkeit des nanopartikulären Eisens wird nun zum Vorteil. Die zu geringe Resorption auf oralem Wege lässt sich durch parenterale Gabe beheben, die bei diesem Verfahren bei normalen Eisensalzen zu hohe Toxizität tritt bei dem weniger verfügbaren Eisen der Partikel nicht auf. Lediglich die erhöhte Infektionsanfälligkeit durch die Bereitstellung von Eisen bleibt ein Problem (siehe oben).
Sichere Aussagen über die Struktur nanoskaliger Oxoeisen(iii)-Partikel sind nur schwer zu erhalten. Eine neuere Arbeit kommt zu dem Schluss, dass das Baumotiv des Akaganéits vorliegen soll [ferritin10]. Ein Vergleich der dieser Aussage zugrunde liegenden Röntgenbeugungsdiagramme (Figure 3 und Figure 4 in [ferritin10]) mit dem Pulverdiagramm von kristallinem Akaganéit (Figure 3 in [ferritin8]) und Beugungsdiagrammen von Ferrihydrit (Figure 2 in [ferritin5]) zeigt die Unsicherheit solcher Interpretationen.
Es gibt nur eine einzige Einkristallstrukturanalyse an einem kohlenhydrat-stabilisierten Oxoeisen(iii)-Nanopartikel. Der Durchmesser von ca. 1.6 nm stimmt mit dem Ergebnis der Lichtstreuung überein, so dass wohl ein typisches Partikel der Lösung kristallisiert werden konnte. Das Übersichtsbild zeigt Kern/Schale-Aufbau (blau: Eisen, grün: Calcium, grau: Kohlenstoff und Wasserstoff, rot: Sauerstoff):
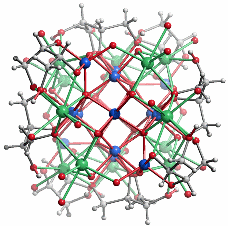
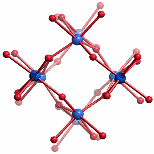
Zehn von insgesamt 14 Eisen(iii)-Zentren bilden einen kompakten Kern, der das Baumuster des Rutils zeigt:
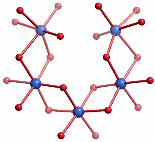
Alternativ lässt sich der innere Kern im Sinne zweier Hämatit-Fragmente interpretieren, die auf symmetrie-unverträgliche Weise ineinandergesteckt sind, so dass weder das Hämatit- noch das Rutil-Baumuster zu einem makroskopischen Kristall weiterwachsen kann; die Abbildung zeigt eines der beiden Hämatit-Fragmente:
[4Fe-4S]-Ferredoxin-Zentren durch Selbstorganisation? Modelle: ja, mitochondriale Ferredoxine werden jedoch durch Metallchaperone unter strikter Kontrolle biosynthetisiert.
Idee: die ersten „biochemischen“ Reaktionen fanden an Eisensulfiden statt.
Aktuelle Ergebnisse: [fes3] zeigt die Bildung von NH3 aus N2 und H2S, treibende Kraft ist die Pyrit-Bildung.
[fe4] zeigt die Bildung carbonylierter Produkte aus einfachen Edukten wie Alkylthiolen und CO unter FeS-Katalyse.
Zu den von Eisensulfid ausgehenden Versuchen erregen derzeit Strukturanalsen Aufsehen, welche die Palette reaktiver Metall-Schwefel-Cluster über die Ferredoxine und über die Nitrogenase-Cluster hinaus erweitern.
Ein Beispiel ist Carboxidothermus-hydrogenoformans-CO-Dehydrogenase (PDB-Code 1SU6).
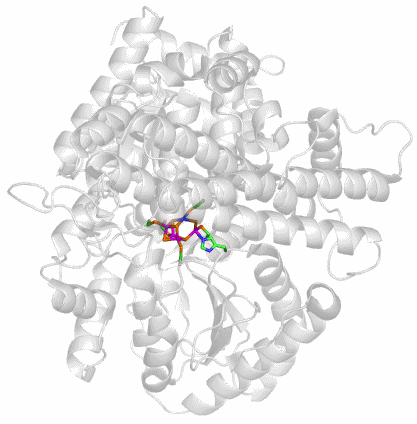
Das aktive Zentrum ist ein ungewöhnlicher [4Fe-Ni-5S]-Cluster, der über vier Cys und ein Fe-gebundenes His mit dem Protein verbunden ist:
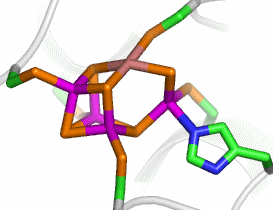
Der Cluster enthält ein quadratisch-planar koordiniertes Nickelatom. Der Mechanismus der CO2-Reduktion ist unbekannt.
Verwandte Zentren sind in [fes2] zusammengestellt, darunter der Acetyl-CoA-erzeugende A-Cluster von CODH/ACS und die Nitrogenase-Cluster.
Solche Strukturen laden zum Vergleich mit Fragmenten von Metallsulfidstrukturen ein, zum Beispiel mit nickelhaltigem Greigit:
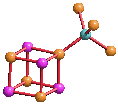
[fes1] erörtert, dass die notwendigen Reaktionsbedingungen in geologischer Umgebung vorliegen.
K. Zeth, S. Offermann, L. O. Essen, D. Oesterhelt: Iron-oxo clusters biomineralizing on protein surfaces: Structural analysis of Halobacterium salinarum DpsA in its low- and high-iron states. Proc. Nat. Acad. Sci. USA 2004, 101, 13780–13785 [ferritin1].
B. L. d’Estaintot, P. Santambrogio, T. Granier, B. Gallois, J. M. Chevalier, G. Précigoux, S. Levi, P. Arosio: Crystal Structure and Biochemical Properties of the Human Mitochondrial Ferritin and its Mutant Ser144Ala. J. Mol. Biol. 2004 340, 277–293 [ferritin2].
H. M. Baker, B. F. Anderson, E. N. Baker: Dealing with iron: Common structural principles in proteins that transport iron and heme. Proc. Natl. Acad. Sci. 2003, 100, 3579–3583 [ferritin3].
X. Liu, E. C. Theil: Ferritins: Dynamic Management of Biological Iron and Oxygen Chemistry. Acc. Chem. Res. 2005, 38, 167–175 [ferritin4].
E. Jansen, A. Kyek, W. Schäfer, U. Schwertmann: The structure of six-line ferrihydrite. Appl. Phys. A 2002, 74, S1004–S1006 [ferritin5].
N. D. Chasteen, P. M. Harrison: Mineralization in Ferritin: An Efficient Means of Iron Storage. J. Struct. Biol. 1999, 126, 182–194 [ferritin6].
C. Quintana, J. M. Cowley, C. Marhic: Electron nanodiffraction and high-resolution electron microscopy studies of the structure and composition of physiological and pathological ferritin. J. Struct. Biol. 2004, 147, 166–178 [ferritin7].
J. E. Post, P. J. Heaney, R. B. v. Dreele, J. C. Hanson: Neutron and temperature-resolved synchrotron X-ray powder diffraction study of akaganéite. Am. Miner. 2003, 88, 782–788 [ferritin8].
K. Ståhl, K. Nielsen, J. Jiang, B. Lebech, J. C. Hanson, P. Norby, J. van Lanschot: On the akaganéite crystal structure, phase transformations and possible role in post-excavational corrosion of iron artifacts. Corrosion Science 2003, 45, 2563–2575 [ferritin9].
D. S. Kudasheva, J. Lai, A. Ulman, M. K. Cowman: Structure of carbohydrate-bound polynuclear iron oxyhydroxide nanoparticles in parenteral formulations. J. Inorg. Biochem. 2004, 98, 1757–1769 [ferritin10].
M. J. Russell, W. Martin: The rocky roots of the acetyl-CoA pathway. Trends Biochem. Sci. 2004, 29, 358–363 [fes1].
C. L. Drennan, J. W. Peters: Surprising cofactors in metalloenzymes. Current Opinion in Structural Biology 2003, 13, 220–226 [fes2].
M. Dörr, J. Käßbohrer, R. Grunert, G. Kreisel, W. A. Brand, R. A. Werner, H. Geilmann, C. Apfel, C. Robl, W. Weigand: Eine mögliche präbiotische Bildung von Ammoniak aus molekularem Stickstoff auf Eisensulfidoberflächen. Angew. Chem. 2003, 115, 1579–1581 [fes3].
G. D. Cody, N. Z. Boctor, T. R. Filley, R. M. Hazen, J. H. Scott, A. Sharma, H. S. Yoder Jr.: Primordial Carbonylated Iron-Sulfur Compounds and the Synthesis of Pyruvate. Science 2000, 289, 1337–1340 [fes4].
K. N. Raymond, E. A. Dertz, S. S. Kim: Enterobactin: An archetype for microbial iron transport. PNAS 2003, 100, 3584–3588 [magnetite1].
In der belebten Natur kommen amorphe Kieselsäureformen in beträchtlicher Menge vor, während Silicate als Biominerale keine Rolle spielen. Am besten untersucht sind Kieselalgen (Diatomeen und Radiolarien), genetesch gut untersucht sind verkieselnde Schwämme. In beiden Gruppen wurden Proteine isoliert, die mit der Polykondensation von Kieselsäure assoziiert sind – die Silaffine bei den Diatomeen, die Silicateine bei den Schwämmen. Die Verkieselung von Pflanzen wie Schachtelhalmen, vor allem aber den Gräsern, ist seit langem bekannt, es gibt jedoch kaum Erkenntnisse über die Einzelschritte der SiO2-Biomineralisation in Pflanzen.
Kieselalgen (engl. diatoms) besitzen ein aus zwei „Theken“ bestehendes Exoskelett, von dessen beiden Schalen bei der vegetativen Zellteilung jeweils eine an jede Tochterzelle weitergegeben wird und jeweils eine neue kleinere Schale (die „Hypotheka“) synthetisiert wird (Fig. 2 in [silica1]). Aufgrund dieses Mechanismus kommt es zur „Diminution“ – ein Teil der Population wird immer kleiner. Kieselalgen korrigieren diesen Vorgang durch geschlechtliche Vermehrung, bei der Theken in arttypischer Größe vollständig neu gebildet werden.
Die Theken sind nicht dicht, sondern sie erlauben den Stoffaustausch aufgrund ihres hierarchischen, porösen Aufbaus (Fig. 1 in [silica1]). Die Bildung des hierachischen Musters ist auf der mikroskopischen Größenskala in den Grundzügen verstanden: Kieselsäure wird in Vesikeln akkumuliert („silica deposition vesicles“, SDVs), in denen ein nicht näher bekannter „Cofaktor“ die Polykondensation trotz der hohen Si-Konzentration verhindert. Die kugelförmigen Vesikel bilden eine Monolage dichtest gepackter SDVs. In diese Form hinein wird Polykieselsäure ausgegossen. Die Vesikel bilden nun eine Tochtergeneration kleinerer Vesikel innerhalb jeder Wabe aus dem ersten Schritt. Anschließend wiederholt sich der Vorgang so oft mit kleineren Vesikel-Generationen wie die Schale Hierarchiestufen aufweist (Fig. 6 in [silica1]).
Auf biochemischer Ebene gibt es Kenntnisse über kieselsäurekondensierende Proteine, den Silaffinen, während der mit diesen zusammenspielende, polykondensations-verhindernde „Cofaktor“ dagegen bisher nicht bekannt ist.
Silaffine lassen sich aus Kieselalgenschalen durch auflösen der anorganischen Komponente isolieren (Fig. 3 in [silica1]). Silaffine sind posttranslational veränderte Proteine, die reich an Lysin (Lys, Einbuchstabencode: K) und Serin (Ser, S) sind. Durch Phosphorylierung vor allem von Ser, aber auch an γ-hydroxyliertem Lys, sowie durch Methylierung und Aminoalkylierung von Lys entsteht ein zwitterionisches Protein (Fig. 4 in [silica1]). Schonend isoliertes Silaffin-1A (natSil-1A) katalysiert die Kieselsäure-Polykondensation in vitro, wobei sphärische Partikel gebildet werden (Fig. 7 in [silica1]).
Sowohl bei den verschiedenen SiO2-Modifikationen als auch bei den Silicaten ist die tetraedrische Umgebung von Silicium die Regel, von der es nur wenig Ausnahmen gibt. Bei SiO2 ist die Ausnahme die Hochdruckmodifikation Stishovit, der in Einschlagskratern von Meteoriten gefunden wurde und der in der Rutil-Struktur kristallisiert. Es wird daher davon ausgegangen, dass amorphe Kieselsäureformen stets tetraedrisch koordiniertes Silicium enthalten.
Kristalline Silicate haben nach dem derzeitigen Kenntnisstand in Organismen keine Bedeutung. Unter den nichtkristallinen Kieselsäuren haben Opale einen engen Bezug zu biogenem SiO2. Opale haben die Formel SiO2 · x H2O. Unterhalb eines Wassergehaltes von ca. 7 % sind Opale hart.
Zur Beschreibung des Verknüpfungsgrades von Siliciumzentren hat sich eine Nomenklatur eingebürgert, bei der die Zahl der nächsten Silicium-Nachbarn um das betrachtete Atom herum einem „Q“ als Hochzahl angefügt wird: ortho-Kieselsäure ist eine Q0-Spezies, Dikieselsäure enthält zwei Q1-Zentren, in linearer Trikieselsäure liegt die Abfolge Q1-Q2-Q1 vor, Cyclotrikieselsäure enthält nur Q2-Bausteine. Quarz besteht aus Q4-Baueinheiten.
Nach dem derzeitigen Kenntnisstand nutzen Kieselalgen die in natürlichen Wässern gelöste ortho-Kieselsäure, H4SiO4. ortho-Kieselsäure ist eine schwache Säure, deren pKA mit ca. 9–10 angegeben wird. Bei physiologischem pH liegt also die undissoziierte Säure vor. Übersteigt die Konzentration der Kieselsäure die Löslichkeit amorpher kondensierter Formen von ca. 100 ppm entsprechend ca. 1 mm, so beginnt die Polykondensation. Die pKA-Werte der Silanol-Funktionen an den verschiedenen Qn-Spezies hängt vom Kondensationsgrad ab. Q0-, Q1- und Q2-gebundene Hydroxylgruppen sind so schwach sauer, dass sie bei physiologischem pH nicht protolysiert sind. pKA-Werte für Q3-Silanolgruppen sind kleiner als 7. In [Shaw 2005] wird der Q3-pKA bei 4–5 gesehen, der Wert für Q2 bei 8–9. In neutraler Lösung wird die Q3-Bildung durch den Energiegewinn bei der Dissoziation von Q3-Spezies vorangetrieben, während sich oberhalb eines pH-Wertes von ca. 11 die verschieden stark kondensierten Silicate nebeneinander beobachten lassen. Analog lässt sich die Verteilung über zahlreiche Spezies in saurer Lösung beobachten, wo nur nicht-protolysierte Kieselsäuren vorliegen. Der pKA-Wert kondensierter kolloidaler Partikel wird mit wenig unter 7 angegeben.
Die einzelnen Stufen der Polykondensation lassen sich bei Kieselsäuren nicht fassen oder gar geplant synthetisieren wie bei den Polyphosphorsäuren (wie würden Sie gezielt Pentanatriumtriphosphat herstellen?). Strukturinformationen stammen vor allem aus zwei Quellen: (1) In Silicaten können die Anionen der Kieselsäuren isoliert und charakterisiert werden. Die größeren nicht-polymeren Oligosilicat-Ionen geben dabei einen Eindruck vom Aufbau kleiner Polykieselsäurepartikel in einer wässrigen Lösung. Das folgende Bild zeigt als Beispiel ein nur aus Q3-Einheiten aufgebautes Dodekasilicat (nach Liebau ein Doppelsechserring-Silicat), das zu den größten wohldefinerten Teilchen gehört, die durch 29Si-NMR-Spektroskopie in wässrig-alkalischen Lösungen nachweisbar sind. Typisch für Oligo- und Polysilicate ist die ausschließliche Eckenverknüpfung der SiO4-Tetraeder.
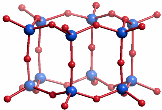
Ein anderes hochsymmetrisches Oligosilicat, das nur aus Q3-Einheiten besteht, ist das Doppelviererring-silicat Si8O208−, das gemäß Si8(μ-O)12(O−)8 würfelförmigen Aufbau hat. (2) Denkt man sich die terminalen O−-Funktionen in dieser Heterocuban-Struktur durch H-Atome ersetzt, so wird ein Silsesquioxan erhalten. Sowohl die Silsesquioxane selbst als auch Derivate, in denen die H-Atome zum Beispiel durch Alkylreste ersetzt sind, sind in großer Zahl isoliert und strukturell untersucht worden. Die erhaltenen Strukturen können als Modelle für Polykieselsäuren angesehen werden. Auch hier ist das Ergebnis, dass die tetraedrischen Baueinheiten stets nur eckenverknüpft vorkommen.
Bei der in-vitro-Messung von zum Beispiel der Aktivität einer Silaffin-Fraktion sollte im Idealfall von ortho-Kieselsäure ausgegangen werden, die jedoch als Reinstoff nicht verfügbar ist. Die übliche Technik besteht in der Verwendung hydrolyseinstabiler Kieselsäureester als Si-Quelle. Am häufigsten werden Tetramethoxysilan (TMOS) und Tetraethoxysilan (TEOS) verwendet. Dabei tritt jedoch eine Schwierigkeit auf, die auf der anderen Seite zu interessanten Schlussfolgerungen führt. Die Hydrolyseempfindlichkeit ist nämlich nicht sehr stark ausgeprägt [silica4], so dass die Reaktivität von zum Beispiel SiCl4 bei Weitem nicht erreicht wird. Perry kritisiert in [silica2] die sich über Minuten und Stunden hinziehende Kieselsäureester-Hydrolyse und verwendet für ihre eigene Studie zur Aminosäure/Kieselsäure-Wechselwirkung ein Tris(catecholato)silicat/Säure-Gemisch, um ohne Verzögerung ortho-Kieselsäure in einer wässrigen Lösung bereitzustellen.
Die Ursache der langsamen und schrittweisen Hydrolyse für die Suche nach bislang unerkannten „Cofaktoren“ aufschlussreich: die Reaktion ist keineswegs kinetisch besonders gehemmt, es fehlt offensichtlich vielmehr die thermodynamische Triebkraft. So konnte Kinrade zeigen, dass in einer alkalischen Wasser/Methanol-Mischung Silicat teilweise zu Methylsilicat reagiert. Si-O-C-Verknüpfungen sind also keineswegs so hydrolyseempfindlich wie dies lange angenommen worden ist, so dass die Suche nach Bioliganden für die Siliciumkomplexierung nicht aussichtslos erscheint.
Im Tierreich ist Verkieselung bei den Schwämmen von besonderer Bedeutung. Schwämme bilden SiO2-Skelettbestandteile aus, die wesentlich härter, dichter und schwerer löslich sind als Kieselalgenschalen. Neben den auffälligen Skelettnadeln (engl. spicules) treten kunstvoll geformte SiO2-„Klammern“ auf, die beim Aufbau des Skeletts als Verbinder dienen. [silica3] zeigt die Formenvielfalt der von Schwämmen hergestellten Bauelemente, deren Funktion nicht allein der Aufbau eines stützenden Skelettes ist.
Die Silicifizierung geht von einem axialen Filament aus, einem Protein, das die Verkieselung steuert. Die Kieselsäureablagerung beginnt mit dem Einfüllen nanometergroßer Kieselsäurekugeln („Nanokugeln“), die dabei zu größeren Kugeln zusammentreten. Fig. 21 in [silica3] zeigt zwei Skelettbausteine in verschiedenem Verkieselungszustand. Die linke der beiden „Klammern“ hat eine erkennbar partikuläre Struktur, die bei der rechten, älteren Klammer durch amorphe Kieselsäure verkittet ist und dadurch glatt wirkt. Die Funktion solcher Verbindungsklammern (Mikroskleren) besteht in der Verbindung der Skelettnadeln (Makroskleren). Dem sich daraus ergebenden flexiblen Skelettaufbau (vgl. Fig 29F in [silica3]) steht in anderen Schwämmen ein festgefügtes SiO2-Skelett gegenüber (Fig 4 in [silica3]).
Das Protein des axialen Filaments in Schwamm-Skelettnadeln lässt sich durch Flusssäurebehandlung vom SiO2 befreien. Es werden zwei Hauptkomponenten gefunden, α- und β-Silicatein. Die Sequenzanalyse zeigt die Verwandtschaft der Silicateine mit den Enzymen der Cathepsin-Familie – Proteasen, die in den Osteoclasten für den Kollagenabbau zuständig sind. Es liegt keine Strukturanalyse vor. Aus Modellierungsstudien wird geschlossen, dass vor allem Serin-Seitenketten, unterstützt von Histidinresten als Base, ein Siliciumzentrum nukleophil angreifen können und so die Polykondensation (und auch die Hydrolyse von Kieselsäurestern) katalysieren. Eine in-vitro-Aktivität von Aminosäuren auf die Kieselsäure-Polykondensation ist dabei nicht überzeugend nachzuweisen [silica2].
Auch die biochemische Seite der Silicateinwirkung ist kaum verstanden. Es scheint ein Zusammenspiel mit Ferritin vorzuliegen, das als innerer Kern des axialen Filament gesehn wird (Fig. 9 in [silica5]).
Isoliertes Silicatein katalysiert die Kieselsäure-Polykondensation, die typischen Aminosäuren des Silicateins und kurzkettige Lysin-Oligomere tun dies nicht. Diese Ergebnisse führen derzeit zu zwei verschiedenen gedanklichen Ansätzen, zum Beispiel SiO2-Kugeln nach dem Vorbild der Skelettnadel-Baueinheiten zu synthetisieren. Der erste Weg nutzt das Protein selbst. So wurde Silicatein mit dem Ziel auf Oberflächen immobilisiert, diese mit strukturiertem SiO2 belegen (Fig. 3 b und c in [silica6]). Die Versuche reichen über SiO2 hinaus. Ein dem Silicatein ähnliches Protein wurde in Pilzen gefunden (die nicht verkieseln!), und für die Synthese von ZrO2-Nanopartikeln genutzt (Fig. 1 in [silica7]).
Eine zweite Idee betrifft die katalytische Wirkung funktioneller Gruppen des Silicateins oder der Silaffine. In [silica8] wird über die Katalyse der Kieselsäure-Polykondensation berichtet, wenn zahlreiche kleinmolekulare, bifunktionelle Verbindungen untersucht werden. Als wirksamer Katalysator stellt sich dabei Cysteamin heraus (Figs. 1 und 4 in [silica8]), bei dem man die Seitenketten von Lysin und Cystein kombinert sehen kann – wenn man denn alles aus dem biochemischen Blickwinkel sehen will.
Was sind konkret die Ziele bei den Versuchen, sphärische SiO2-Partikel herzustellen. Ein besonders interessantes Ziel ist die biomimetische Synthese neuer Opal-Varianten. Opale, die eine kubisch-flächenzentrierte Anordnung sub-μm-großer hydratisierter SiO2-Partikel darstellen, haben eine Fülle von Anwendungen [silica9], siehe dort Fig. 1 für eine Abbildung des Opal-Aufbaus. Eine besonders interessante Verwendung ist die Nutzung als photonische Kristalle [silica11]. Für deren Aufbau ist außer den SiO2-Kugeln das umgekehrte Prinzip genutzt worden, nämlich der Synthese eines inversen Opals, dessen Konstruktion eine weitere Anwendung der Kieselsäure-Polykondensation ist (Fig. 6 in [silica10], [silica11].
Üblicherweise wird ortho-Kieselsäure als die Transportform des Siliciums angesehen. Es ist daher bemerkenswert, das Schwämme über ein Silicase genanntes Enzym verfügen. Auch für dieses sind keine Strukturdaten bekannt, es wird jedoch als der Carboanhydrase-Familie zugehörig beschrieben.
Nicht nur Moose, Bärlappe, Schachtelhalme, Farne und Sauergräser sind verkieselnde Pflanzen, sondern auch die wichtigen Nutzpflanzen aus der Familie der Süßgräser. Neben den bei uns üblichen Getreidearten sind dies vor allem Reis und Bambus. Der Siliciumgehalt solcher Pflanzen liegt im Bereich einiger Prozent (man denke an die Bezeichnung „Zinnkraut“ für Schachtelhalm, die an die Verwendung des getrockneten Krauts als Scheuermittel für Zinngeschirr erinnert).
M. Sumper, N. Kröger: Silica formation in diatoms: the function of long-chain polyamines and silaffins: J. Mater. Chem. 2004, 14, 2059–2065 [silica1].
D. Belton, G. Paine, S. V. Patwardhan, C. C. Perry: Towards an understanding of (bio)silicification: the role of amino acids and lysine oligomers in silicification. J . Mater. Chem. 2004, 14, 2231–2241 [silica2].
M.-J. Uriz, X. Turon, M. A. Becerro, G. Agell: Siliceous Spicules and Skeleton Frameworks in Sponges: Origin, Diversity, Ultrastructural Patterns, and Biological Functions. Micr. Res. Techn. 2003, 62, 279–299 [silica3].
F. Brunet, B. Cabane, M. Dubois, B. Perly: Sol-Gel Polymerization Studied through 29Si NMR with Polarization Transfer. J. Phys. Chem. 1991, 95, 945–951 [silica4].
W. E. G. Müller, A. Krasko, G. Le Pennec, H. C. Schröder: Biochemistry and Cell Biology of Silica Formation in Sponges. Micr. Res. Tech. 2003, 62, 368–377 [silica5].
M. N. Tahir, P. Théato, W. E. G. Müller, H. C. Schröder, A. Janshoff, J. Zhang, J. Huthe, W. Tremel: Monitoring the formation of biosilica catalysed by histidine-tagged silicatein. Chem. Comm. 2004, 2848–2849 [silica6].
V. Bansal, D. Rautaray, A. Ahmad , M. Sastry: Biosynthesis of zirconia nanoparticles using the fungus Fusarium oxysporum. J. Mater. Chem. 2004, 14, 3303–3305 [silica7].
K. M. Roth, Y. Zhou, W. Yang, D. E. Morse: Bifunctional Small Molecules Are Biomimetic Catalysts for Silica Synthesis at Neutral pH. J. Am. Chem. Soc. 2005, in press [silica8].
M. R. Newton, A. K. Bohaty, H. S. White, I. Zharov: Chemically Modified Opals as Thin Permselective Nanoporous Membranes. J. Am. Chem. Soc. 2005, 127, 7268–7269 [silica9] .
Z. Zhou, X. S. Zhao: Opal and Inverse Opal Fabricated with a Flow-Controlled Vertical Deposition Method. Langmuir 2005, 21, 4717–4723 [silica10].
J. Ozin: The photonic opal – the jewel in the crown of optical information processing. Chem. Comm. 2003, 2639–2643 [silica11].
E. Bäuerlein (Ed.): Biomineralization: Progress in Biology, Molecular Biology and Application. 2nd Edition. Wiley-VCH 2004