 Der Liebig-Lab-Vorkurs
Der Liebig-Lab-Vorkurs
 Der Liebig-Lab-Vorkurs
Der Liebig-Lab-Vorkurs
Beginn: Dienstag, 20. Oktober 2020.
Ort: Praktikumssäle in Haus D.
Studierende des Bachelorstudiengangs Chemie und Biochemie, die anschließend am Liebig-Laboratorium teilnehmen wollen.
Nach einer kurzen Einführung in den Gebrauch typischen Laborgeräts kommt als erster Schwerpunkt (Abschnitt 2) der Umgang mit wichtigen Gefahrstoffen an die Reihe. Mit Salz-, Schwefel- und Salpetersäure, Ammoniaklösung, Alkalilaugen und Wasserstoffperoxidlösung werden Stoffe aus dem Laboralltag eingeführt, die bei unsachgemäßem Umgang ein beachtliches Risikopotential aufweisen. Hier lernen Sie als Grundlage der Arbeitssicherheit die vom Gesetzgeber geforderten Betriebsanweisungen kennen. In Abschnitt 4 werden Sie sich mit dem zweiten Schwerpunkt befassen, den Säure-Base-Gleichgewichten.
Der Vorkurs zum Liebig-Lab soll Sie (1) mit den handwerklichen Grundlagen des nachfolgenden Liebig-Laboratoriums vertraut machen, (2) das Führen eines Laborprotokolls lehren, (3) mit den charakteristischen Eigenschaften wichtiger Stoffgruppen bekannt machen, und (4) an das Aufstellen von Reaktionsgleichungen heranführen. Der „rote Faden“ des Vorkurses ist der sichere Umgang mit Gefahrstoffen.
Der Ablauf ist der Gleiche wie im nachfolgenden Liebig-Lab: Praktikum von 13:00–17:00 Uhr. Alles, was bearbeitet wurde, bleibt am Platz stehen. Der Gruppenassistent prüft ab ca. 16:00 Uhr stichprobenartig, ob das praktisch Ausgeführte verstanden wurde (ob zum Beispiel Reaktionsgleichungen aufgestellt werden können). Gegen 17:00 Uhr gibt es eine Schlussbesprechung von ca. ½ Stunde im Saal, wo wiederkehrende Schwierigkeiten besprochen werden.
Am Vormittag eines Praktikumstages findet eine begleitende Vorlesung statt.
Prüfen Sie hier, ob Ihr Browser das Skript korrekt darstellt.
Um das Skript auszudrucken, verwenden Sie am Besten die pdf-Version. Studierende, die am Praktikum teilnehmen, erhalten einen Ausdruck bei Praktikumsbeginn.
Betriebsanweisungen bilden den sicherheitstechnischen Rahmen des Praktikums. Dies beginnt für die Arbeit im Vorkurs und im Liebig-Laboratorium mit einer übergeordneten Anweisung.
Diese Allgemeine Betriebsanweisung fasst die Sicherheitsregeln des Praktikums allgemein und knapp zusammen. Sie basiert auf zwei Dokumenten, die zu Beginn der Anweisung als Quellen genannt sind, der GUV-R 120 Laboratorien und der Laboratoriumsordnung des Departments.
Als anschauliche Einführung liegt eine Broschüre der Gesetzlichen Unfallversicherung vor, der unter der Bestellnummer GUV-I 8553 herausgegebenen Einführung für Studierende mit dem Titel Sicheres Arbeiten in chemischen Laboratorien.
Sie erhalten zu Beginn des Praktikums jeweils ein Exemplar der Texte. Die dort angegebenen Regeln sind für Sie bindend. Darüberhinaus lernen Sie stoffbezogene Betriebsanweisungen kennen, in denen auf speziellere Gefahren eingegangen wird.
Das erste Hauptziel des Vorkurses, mit Gefahrstoffen sicher umzugehen, soll durch Versuche erreicht werden, bei denen die Warnungen der Betriebsanweisungen hinterfragt und verstanden werden. Behandelt werden die oben genannten wichtigsten Gefahrstoffe des ersten Semesters: Salz-, Schwefel- und Salpetersäure, Ammoniaklösung, Natron- und Kalilauge sowie Wasserstoffperoxid. Hinzu kommen wichtige Laborgase. Anschließend folgt das zweite Hauptziel des Vorkurses, nämlich eine Einführung in die Maßanalyse, die im Mittelpunkt des nachfolgenden Liebig-Laboratoriums stehen wird.
Das Arbeiten mit gefährlichen Stoffen, vor allem mit giftigen oder brennbaren Gasen und Dämpfen, ist nur in einem gut ziehenden Abzug erlaubt. Die Abzugsleistung ist im Wesentlichen davon abhängig, wie weit der Frontschieber des Abzugs geöffnet ist. Bei weit geöffnetem Frontschieber verringert sich die Wirksamkeit des Abzuges erheblich. Gehen Personen am geöffneten Abzug vorbei, kann es zu Verwirbelungen und in deren Folge zum Ausbruch von Stoffen aus dem Abzug kommen. Halten Sie daher die Frontscheibe immer so weit wie möglich geschlossen und benutzen Sie zum Arbeiten nach Möglichkeit die seitlich verschiebbaren Arbeitsfenster. Neben dem Abführen von Gasen und Dämpfen dient der Abzug auch als Schutz vor verspritzenden Substanzen und als Splitterschutz. Trotzdem haben Sie, wie bei allen Arbeiten im Labor, auch beim Arbeiten am Abzug eine Schutzbrille zu tragen.
Wird am Abzug nicht gearbeitet, sind sowohl der Frontschieber als auch die Arbeitsfenster immer geschlossen zu halten! Der Abzug darf nicht als Lagerplatz verwendet werden. Entfernen Sie daher nicht benötigte Chemikalien oder Geräte aus dem Abzug und stellen Sie diese an die dafür vorgesehenen Lagerplätze. Schauen Sie nun, wie sich diese Erfahrungen in einer gerätebezogenen Betriebsanweisung niedergeschlagen haben.
Zum Erhitzen wird im Labor gewöhnlich der Bunsenbrenner benutzt. Im unteren Teil enthält er eine Düse, aus der Erdgas (Methan) ausströmt, und eine Vorrichtung, um Luft in verschiedenen Mengen in das Brennerrohr einzulassen. Ohne Luftzufuhr erhält man eine leuchtende Flamme. Ein Teil des Methans wird zunächst nur zu Kohlenstoff und Wasser oxidiert; bei der Flammentemperatur leuchten die gebildeten festen Kohlenstoff-(Ruß-)Teilchen. Leicht Sauerstoff abgebende Substanzen werden in der leuchtenden Flamme reduziert.
Bei Luftzutritt verbrennt Erdgas vollständig zu Kohlendioxid und Wasser. Bei der entstehenden „nichtleuchtenden“ Flamme lässt sich ein innerer, blau leuchtender Kegel erkennen, in dem reduzierende Bedingungen herrschen. Um diesen Kegel herum liegen oxidierende Bedingungen vor. An der Spitze des blauen Kegels ist die heißeste Stelle der Flamme:

Ist die Luftzufuhr zu groß oder der Gasdruck zu klein, so schlägt der Brenner durch, dass heißt, das Gas brennt im Innern des Brennrohres an der Gasaustrittsdüse. In diesem Fall muss die Gaszufuhr sofort abgestellt werden. Nach dem Erkalten des Brenners stellt man dann die Luftzufuhr etwas kleiner oder vergrößert die Gaszufuhr.

Waagen müssen stets sauber gehalten und schonend behandelt werden. Zunächst wird ein Wägegefäß auf die Waage gestellt (niemals Substanzen direkt auf die Waage geben!), dann wird „Tara“ gedrückt, um das Gefäß nicht mitzuwiegen. Nach dem Tarieren wird die zu wägende Substanz abgewogen. Als Wägegefäß für Feststoffe dient ein Uhrglas, Wägeschiffchen oder ein anderes leichtes(!) Gefäß. Im Falle einer hygroskopischen, flüssigen oder leicht flüchtigen Substanz ist ein Gefäß mit eingeschliffenem Deckel zu verwenden. Nun kann die Wägung vorgenommen werden. Informieren Sie sich vor Beginn der Wägung anhand der Anleitung über die Bedienung der Waage.
Finden Sie mit Hilfe der Bedienungsanleitung heraus, warum Sie nicht irgend ein beliebig schweres Wägegefäß verwenden dürfen – schließlich tariert man doch.
Im Labor wird überwiegend mit Glasgeräten gearbeitet, weshalb Sie die einfachsten Arten der Glasbearbeitung beherrschen sollten, auch um kleinere Beschädigungen schnell zu beheben.
Das Rohr wird mit einem scharfen Glasmesser zu einem Viertel seines Umfanges an der gewünschten Stelle eingeritzt. Durch leichtes Ziehen und Biegen gegen die Kerbe wird das Glasrohr auseinandergesprengt.
Zerteilte und abgesprengte Glasrohre oder Stäbe sind an den Schnittkanten scharf. Ihre Enden müssen daher rundgeschmolzen werden. In der leuchtenden Flamme wird das Rohrende unter gleichmäßiger Drehung erwärmt, anschließend wird bei entleuchteter Flamme erhitzt, bis die Kanten erweichen. Je weiter das Rohr ist, desto vorsichtiger muss die Erwärmung vor sich gehen. Zum Schluss wird in der leuchtenden Flamme getempert.
Um ein Rohr zu biegen, wird eine Seite mit einem Stopfen verschlossen. Anschließend wird unter gleichmäßigem(!) Drehen bis zum Erweichen des Glases erhitzt (wie oben mit leuchtender Flamme beginnen). Die erweichte Stelle wird nach unten weggebogen und eine eventuell entstandene Verengung außerhalb der Flamme vorsichtig wieder aufgeblasen. Zum Schluss wird in der leuchtenden Flamme getempert.
Viele analytische Nachweisreaktionen sind sehr empfindlich, sie sprechen auf kleinste Substanzmengen an. Das bringt es mit sich, dass nachlässig gereinigtes Laborgerät zur Fehlinterpretation von Versuchen führt. Der folgende Versuch regt Sie dazu an, einen Spülvorgang als mehrfaches Verdünnen um einen charakteristischen Faktor zu begreifen.
Dieser Versuch zeigt sehr schön, dass ein Laborgefäß nach dem Ausspülen „sauber“ aussieht, es aber nicht immer ist.
In drei Reagenzgläser werden je 2 mL einer Eisen(III)-chloridlösung (ca. 0,1 mol L−1) gegeben. Nach dem Ausgießen der Lösung spült man das erste Glas mit 1 mL Wasser aus. Das zweite Glas wird zweimal mit je 1 mL Wasser ausgespült. Das dritte Glas wird unter Verwendung von Reagenzglasbürste und Spülmittel/Wasser gründlich gereinigt und anschließend mehrmals mit destilliertem Wasser ausgespült. Durch Zusatz von Ammoniumthiocyanat-Lösung (ca. 0,1 mol L−1) lassen sich noch vorhandene Eisen(III)-Ionen durch eine Rotfärbung nachweisen. Dies sollte bei den ersten beiden Reagenzgläsern der Fall sein, jedoch nicht bei dem sorgfältig gereinigten Glas.
Gehen Sie davon aus, dass beim Ausgießen immer 1 Tropfen im Glas zurückbleibt. Das Volumen eines Wassertropfens ist ungefähr 0,05 mL. Berechnen Sie die Eisen(III)-Konzentration in mol L−1 nach dem ersten und nach dem zweiten Wiederauffüllen mit je 1 mL Wasser.
Reagenzgläser werden für chemische Reaktionen und zur Aufbewahrung von kleinen Flüssigkeitsmengen verwendet. Zur Lagerung oder zum Durchmischen werden sie mit einem Kork- oder Kunststoffstopfen verschlossen (niemals die Öffnung mit dem Daumen verschließen). Gläser, die in einer Flamme erwärmt werden sollen, sind meist dünnwandig, um Bruch durch thermische Spannungen zu vermeiden.
Einmal richtig und einmal falsch. Wenn Sie beides gesehen haben, werden Sie es nicht mehr falsch machen. Ziel ist, dass Sie einen Siedeverzug kennenlernen und eine Möglichkeit, diesen zu vermeiden.
Ein Reagenzglas wird etwa zur Hälfte mit Wasser gefüllt und mit Hilfe einer Reagenzglasklammer senkrecht in die nichtleuchtende Bunsenbrennerflamme gehalten. Innerhalb kurzer Zeit errreicht das Wasser Siedetemperatur. Es kommt in der Regel zu einem Siedeverzug, der den Inhalt des Reagenzglases herausschiessen lässt; Reagenzglas nicht auf den Nachbarn richten!
Ein zweiter Versuch zeigt, wie ein solcher unerwünschter Siedeverzug vermieden wird. Das Reagenzglas wird nur etwa zu ¼ mit Wasser gefüllt. Dann wird es schräg in die nichtleuchtende Bunsenbrennerflamme gehalten und dabei geschüttelt. Das Wasser sollte nun gleichmäßig sieden und nicht wie vorher herausspritzen, da eine lokale Erhitzung vermieden wird.
Beschreiben Sie, wie es zu einem Siedeverzug kommen kann.
Es geht um Fingerfertigkeit. Wenn Sie Mühe haben, kleine Substanzmengen so sicher zu dosieren, dass nichts überschäumt, wiederholen Sie den Versuch mehrmals.
In einem Reagenzglas wird ca. 1 mL gesättigte Natriumcarbonat-Lösung mit 2 Tropfen Methylrot-Indikatorlösung versetzt. Nun wird tropfenweise verdünnte Salzsäure hinzugegeben und durch Schütteln des Reagenzglases gemischt. Es wird weitere Säure zugegeben, bis der Indikator umschlägt.
Welches Gas entwickelt sich bei der Säurezugabe? Formulieren Sie eine Reaktionsgleichung.
Besonders wichtig: die üblichen Konzentrationsmaße. Die Stoffmengenkonzentration oder Molarität (c) wird definiert als Stoffmenge (n) pro Volumen (V), Einheit: mol L−1, abgekürzt m.
Der meist in Prozent angegebene Massenanteil („Massenprozent“) ist definiert als Masse pro Gesamtmasse des Gemisches, Einheit: 1 oder Prozent. 30%ige Salzsäure enthält daher 30 g HCl in 100 g Salzsäure.
Die Dichte von 30%iger Salzsäure ist 1,15 kg L−1. Wieviel mL dieser Säure müssen abgemessen werden, wenn 36,5 g HCl benötigt werden?
Die meisten Versuche werden in Lösung durchgeführt. Das Gelingen des Versuches hängt oft von der geeigneten Konzentration der Lösung ab. Arbeitet man mit zu konzentrierten Lösungen, so können Konzentrationsniederschläge auftreten oder aber die Niederschlagsmenge ist so groß, dass die charakteristische Form der Abscheidung (kristallin, flockig, gelartig) nicht oder nur schwer erkennbar ist. In zu verdünnten Lösungen kann eine Reaktion ganz ausbleiben (Grenzkonzentration, Erfassungsgrenze) oder mit großer zeitlicher Verzögerung eintreten.
Versetzen Sie eine kleine Probe einer 5%igen Eisen(III)-chloridlösung in einem Reagenzglas mit wenig konzentrierter Ammoniaklösung; eine zweite Probe der Lösung wird auf das 20-fache verdünnt und ein Teil der Probe mit konzentrierter Ammoniaklösung versetzt. Schließlich versuchen Sie, wie weit die Lösung verdünnt werden muss, damit mit Ammoniaklösung keine deutliche Fällung mehr auftritt.
Formulieren sie die Gleichung für die Reaktion von Eisen(III)-Ionen mit Ammoniaklösung.
Eine Vorübung zu Abschnitt 3:
Stellen Sie eine Konzentrationsreihe aus einer 10 m HCl mit folgenden Konzentrationen her: 0,1 m, 0,001 m, 0,00001 m. Bestimmen Sie anschließend mit Hilfe von pH-Papier den pH-Wert der Lösungen.
Die einfache Arrhenius-Theorie (Säuren zerfallen [„dissoziieren“] in H+-Ionen und Säurerest-Anionen) erlaubt Ihnen, das Ergebnis einzuordnen. Nehmen Sie an, dass jedes Molekül HCl dissoziiert. Der pH-Wert ist als negativer Zehnerlogarithmus der H+-Konzentration definiert. Rechnen Sie für die drei Konzentrationen die zu erwarteten pH-Werte aus und vergleichen Sie mit Ihrem Ergebnis.
Unter einem Gemisch versteht man einen Stoff, der mindestens aus zwei Reinstoffen besteht. Die spezifischen Eigenschaften wie zum Beispiel Dichte, Siedepunkt oder Farbe sind vom Mischungsverhältnis (Massenverhältnis) der Komponenten abhängig. Man unterscheidet homogene Gemische, bei denen man weder mit dem Auge noch mit dem Mikroskop die Zusammensetzung verschiedener Reinstoffe erkennen kann und heterogene Gemische.
Das zu trennende Dreikomponenten-Gemisch enthält Kupfer(II)-chlorid-Dihydrat, Zimtsäure und Sand. Löslichkeiten in Wasser: CuCl2·2H2O: bei 0 °C 1,10 g mL−1, bei 100 °C 1,92 g mL−1; Zimtsäure: bei 0 °C 0,0004 g mL−1, in der Siedehitze > 1,60 g mL−1 (wenn etwas Ethanol zugesetzt wird); SiO2: praktisch unlöslich.
Zunächst werden 5 g des Gemisches in ein kleines Becherglas gegeben, 10 mL Wasser hinzugefügt und 1 Minute gerührt. Es wird nun abfiltriert. Wenn der Rückstand im Filter fast trocken ist, wird mit ca. 1 mL Wasser gewaschen. Der beschriebene Waschvorgang wird solange wiederholt, bis das ablaufende Filtrat keinen Anteil der gut löslichen Komponente mehr zeigt. Das Filtrat stellt man zunächst (weiter bei •) zur Seite (Welche Komponente befindet sich im Filtrat?).
Das Filterpapier mit dem Rückstand gibt man mit 20 mL Wasser und 10 mL Ethanol in einen Erlenmeyerkolben, wäscht es darin, entfernt das Papier und erhitzt auf der Heizplatte unter Rühren zum Sieden. Dann wird möglichst heiss durch einen erwärmten Trichter filtriert. Der Rückstand im Filter wird zweimal mit je 10 mL heißem Wasser gewaschen. Das Filtrat wird nun auf Zimmertemperatur abgekühlt und man stellt es mindestens 40 Minuten in ein Eiswasser-Bad (weiter bei ••).
Während der Wartezeit wird der Filterrückstand mit Wasser gewaschen und getrocknet. Um welche Komponente handelt es sich? Bestimmen Sie die Masse.
•• Das gebildete Kristallisat wird mit Hilfe einer Nutsche isoliert und gut trocken gesaugt. Die gewonnene Komponente wird zum weiteren Trocknen in den Exsikkator gegeben. Am nächsten Praktikumstag wird der Schmelzpunkt und die Masse bestimmt. Vergleichen Sie Ihren Wert für den Schmelzpunkt mit dem Literaturwert von 134–136 °C. Um welche Komponente handelt es sich?
• Das farbige Filtrat der ersten Trennung wird unter starkem Rühren bis fast zur Trockene eingedampft (am Schluss vorsichtig und nicht zu stark erhitzen). Erscheint wasserfreies Kupfer(II)-chlorid (Farbe!), so beendet man das Heizen. Bestimmen Sie die Masse.
Die Arbeit mit Gefahrstoffen ist in einem chemischen Laboratorium Alltag. In diesem Kapitel steht daher ein Lernziel im Vordergrund: der sichere Umgang mit Gefahrstoffen. Die vom Gesetzgeber geforderte praktische Hilfe hierzu sind die Betriebsanweisungen. Die ersten Betriebsanweisungen hatten Sie oben bereits kennengelernt, nämlich die Allgemeine Betriebsanweisung, die den Rahmen für das sichere Arbeiten im Praktikum bildet und die gerätebezogene Betriebsanweisung für den Umgang mit Laborabzügen. In den stoffbezogenen Betriebsanweisungen dieses Kapitels sind oft Gefahren genannt, ohne dass jedoch deren chemischer Hintergrund immer beleuchtet wird. Es bleibt also die Aufgabe zu verstehen, was bei einer Reaktion geschieht und worin die Gefahr besteht. Sie werden daher solche gefährlichen Reaktionen untersuchen, den Stoffumsatz durch eine Reaktionsgleichung ausdrücken und die Ursache der Gefahr erkennen.
Sie erkennen Versuche, durch welche die Betriebsanweisungen näher beleuchtet werden, an der Gestaltung der Versuchsüberschrift in der (derzeit noch) orangen Warnfarbe und dem nachgestellten Vermerk „(BA!)“:
Die Betriebsanweisungen der in diesem Kapitel verwendeten Ausgangsstoffe sind bereits in diesem Skript verlinkt. Allerdings entstehen bei einigen dieser Versuche weitere Stoffe, von denen eine Gefahr ausgehen kann, beispielsweise giftige oder brennbare Gase. Machen Sie sich also vor der Durchführung der Versuche klar, welche Stoffe entstehen können (Reaktionsgleichung!) und informieren Sie sich gegebenfalls selbstständig anhand von Betriebsanweisungen über mögliche Gefahren. Hierzu steht ein Ordner mit Betriebsanweisungen im Praktikumssaal aus.
Salzsäure gehört zu den wichtigsten Grundchemikalien des Labors. Im menschlichen Organsismus kommt sie als „Magensäure“ in einer Maximalkonzentration von ca. 0,1 mol L−1 vor. Bereits diese Konzentration reicht aus, um bei Refluxerkrankungen („Sodbrennen“) die Speiseröhre zu verätzen. Die im Labor verwendete Salzsäure ist bis zu 100-mal höher konzentriert.
Konzentrierte Salzsäure ist eine ca. 35%ige Lösung des farblosen und stechend riechenden Gases Chlorwasserstoff (HCl) in Wasser, verdünnte Salzsäure ist eine ca. 7%ige Lösung. Wird konzentrierte Salzsäure erhitzt, so destilliert hauptsächlich Chlorwasserstoff und wenig Wasser ab, bis die Konzentration der Lösung auf 20 % gesunken ist. Erhitzt man umgekehrt verdünnte Salzsäure, so entweicht hauptsächlich Wasser, bis die Säure wieder 20%ig ist. Die Dichte einer Salzsäure folgt einer einfachen Faustformel: Division der Prozentangabe durch 200, dann Addition von 1 ergibt die Dichte in g cm−3. 34%ige Salzsäure hat also wegen 34/200 + 1 = 1,17 die Dichte 1,17 g cm−3.
Errechnen Sie die Stoffmengenkonzentration (Molarität) von konzentrierter und verdünnter Salzsäure.
Salzsäure reagiert mit hinreichend starken Reduktionsmitteln heftig unter Wasserstoffentwicklung, während starke Oxidationsmittel Chlor freisetzen. Neben der großen Exothermie der Reaktionen ergeben sich besondere Gefahren aus der Brennbarkeit von Wasserstoff und der Aggressivität von Chlorgas.
Der Umgang mit konzentrierter Salzsäure (ab 25 %) wird durch eine Betriebsanweisung geregelt.
Die Betriebsanweisung zur konzentrierten Salzsäure führt deren Umsetzung mit Aluminium als besondere Gefahr auf. Der Versuch soll aufzeigen, wieso.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Führen Sie die Versuche, bei denen eine Gasentwicklungsapparatur benötigt wird, in Zweiergruppen aus.
Geben Sie ca. 250 mg Aluminium-Grieß in den 100-mL-Einhalsrundkolben, der Ihnen für diesen Versuch zur Verfügung gestellten Gasentwicklungsapparatur. Füllen Sie die beigegebene Kristallisierschale (hier als pneumatische Wanne zu verwenden) mit ausreichend Wasser. Legen Sie darin unter Wasser einen passenden Gummistopfen für ein Reagenzglas bereit, in dem das bei der Reaktion freigesetzte Gas gesammelt werden soll, und tauchen Sie das vollständig mit Wasser gefüllte Reagenzglas in das Wasser der pneumatischen Wanne ein. Versetzen Sie nun im Reaktionskolben das Aluminium mit ca. 10 mL verdünnter Salzsäure. Verschließen Sie rasch den Kolben mit dem Gasüberleitungsrohr, das in einem durchbohrten Stopfen steckt, und tauchen Sie dessen Ende unter die Wasseroberfläche der pneumatischen Wanne. Es setzt langsam Gasentwicklung ein, die nach kurzer Zeit heftiger wird. Beginnen Sie mit dem Auffangen des Gases erst nach ca. zwei Minuten, so dass zunächst die Luft aus dem Reaktionskolben verdrängt wird. Halten Sie dann das Ende des Gaseinleitungsrohres direkt unter das senkrecht aufgerichtete Reagenzglas. Wenn das Wasser im Reagenzglas vollständig verdrängt ist, wird es unter Wasser mit dem Gummistopfen fest verschlossen. Nehmen Sie nun das verschlossene Glas aus dem Wasser. Ziehen Sie dann den Stopfen ab und halten Sie die Öffnung des Reagenzglases sofort in die Flamme des Bunsenbrenners (Öffnung stets nach unten weisen lassen).
Um welches Gas handelt es sich (Reaktionsgleichung)? Warum führt die Betriebsanweisung die Reaktion mit Aluminium als Gefahr auf?
Die Betriebsanweisung zur konzentrierten Salzsäure führt deren Umsetzung mit Kaliumpermanganat als besondere Gefahr auf. Der Versuch soll aufzeigen, wieso.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie ca. 800 mg gepulvertes Kaliumpermanganat in angehäufter Form auf ein Uhrglas. Tropfen Sie anschließend mittels einer Pasteurpipette langsam konzentrierte Salzsäure zu. Dabei entsteht ein Gas. Die Farbe des Gases können Sie deutlicher wahrnehmen, indem Sie das Experiment vor einem weißen Hintergrund (zum Beispiel einem Bogen weißen Papiers) durchführen.
Um welches Gas handelt es sich (Reaktionsgleichung)? Warum führt die Betriebsanweisung die Reaktion mit Kaliumpermanganat als Gefahr auf?
Die Betriebsanweisung zur konzentrierten Salzsäure führt deren Umsetzung mit Salzen von Halogensauerstoffsäuren als besondere Gefahr auf. Der Versuch soll aufzeigen, wieso.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie ca. 500 mg gepulvertes Kaliumchlorat in angehäufter Form auf ein Uhrglas. Tropfen Sie anschließend mittels einer Pasteurpipette langsam konzentrierte Salzsäure zu. Dabei entsteht ein Gas. Die Farbe des Gases können Sie auch hier deutlicher wahrnehmen, indem Sie das Experiment vor einem weißen Hintergrund durchführen.
Um welches Gas handelt es sich (Reaktionsgleichung)? Warum führt die Betriebsanweisung die Reaktion mit Salzen von Halogensauerstoffsäuren als Gefahr auf?
… die Reaktion mit Hydriden (Calciumhydrid, Lithiumaluminiumhydrid, beides starke Trockenmittel), die zu einer explosionsartigen Entwicklung von Wasserstoff führen kann.
Formulieren Sie die Reaktionsgleichung für die Umsetzung von Calciumhydrid mit Salzsäure.
Bei den Versuchen mit Salzsäure ergab sich die Entstehung von Wasserstoff als eine wesentliche Gefahrenquelle. Andererseits gehört Wasserstoff zu den wichtigsten Laborgasen überhaupt. Die sichere Herstellung und Handhabung von Wasserstoff gehört daher zu den Grundoperationen in einem chemischen Laboratorium.
Wasserstoff ist ein farbloses, brennbares und geruchloses Gas, das viel leichter als Luft ist. Es bildet mit Luft, mehr noch mit reinem Sauerstoff oder auch Chlor, explosionsfähige Gemische. Im Laboratorium wird Wasserstoff bei der Einwirkung von Säuren auf Zink oder von Laugen auf Aluminium erhalten, in der belebten Natur produzieren und nutzen Mikroorganismen Wasserstoff durch Hydrogenasen. Während Wasserstoff in Wasser oder in Säuren die Oxidationsstufe +I aufweist, kann er mit Metallen Hydride mit der Oxidationsstufe −I bilden, wie beispielsweise in LiH.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie 2 Zinkgranalien in den Rundkolben der für diesen Versuch bereit gestellten Gasentwicklungsapparatur und versetzen Sie diese mit ca. 5 mL konzentrierter Salzsäure. Fangen Sie das entstehende Gas wie zuvor beschrieben in einem Reagenzglas auf und führen Sie eine Knallgasprobe durch. Möchte man die Wasserstoffentwicklung grob steuern, kann man dies durch dosiertes Zutropfen der Salzsäure unter Verwendung eines Tropftrichters erreichen.
Wieviel Liter Wasserstoffgas lässt sich unter Standardbedingungen aus 100 g Zink und einer ausreichenden Säuremenge gewinnen?
Schwefelsäure ist eine der wichtigsten Grundchemikalien. Die Schwefelsäureproduktion gehört zu den Kennzahlen, die in die Bewertung der Wirtschaftsleistung einer Volkswirtschaft eingehen. Im Gegensatz zu der aus zwei Dritteln Wasser bestehenden konzentrierten Salzsäure ist konzentrierte Schwefelsäure fast wasserfrei.
Konzentrierte Schwefelsäure, eine ölige Flüssigkeit, ist 96%ig. Die chemischen Eigenschaften sind durch zwei Charakteristika geprägt: Schwefelsäure ist (1) außerordentlich wasseranziehend und (2) stark oxidierend. „Wasseranziehend“ bezieht sich dabei unmittelbar auf den Stoff Wasser selbst, darüberhinausgehend aber auch auf komplexe chemische Reaktionen, in deren Verlauf das Reaktionsprodukt Wasser entsteht. Achten Sie hier besonders auf den Versuch mit Ameisensäure. Starke Reduktionsmittel wie Zink reagieren mit heißer konzentrierter Schwefelsäure unter sehr weit gehender Reduktion der Säure zu elementarem Schwefel und in geringem Umfang sogar zu Hydrogensulfid, H2S. Beachten Sie bei den Versuchen den Einfluss der Reaktivität des Metalls und der Konzentration der Säure.
Der Umgang mit Schwefelsäure (ab 5 %) wird durch eine Betriebsanweisung geregelt.
Bei der Herstellung von verdünnter Schwefelsäure wird stets die konzentrierte Säure langsam und unter guter Durchmischung in das Wasser gegossen – nicht umgekehrt: „erst das Wasser, dann die Säure, sonst geschieht das Ungeheure“! Heiße konzentrierte Schwefelsäure darf keinesfalls verdünnt oder in den Ausguss gegossen werden.
Nochmal, auf die Reihenfolge kommt es an: „Erst das Wasser, …“
Zu 3 mL Wasser gieße man aus einem zweiten Reagenzglas etwa den gleichen Raumteil konzentrierter Schwefelsäure. Die Mischung erwärmt sich stark.
Jetzt, wo Sie die freiwerdende Wärmemenge gespürt haben: Was ist denn das „Ungeheure“? Also was genau kann passieren, wenn Wasser in konzentrierte Schwefelsäure gegossen wird?
Das Verkohlen organischen Materials bei Schwefelsäurezusatz ist formal ein Entzug der Elemente des Wassers. Ein Beispiel ist das Verkohlen von Traubenzucker:
C6H12O6 → 6 C + 6 H2O.
Ameisensäure ist einer der seltenen Fälle, bei denen bei der Umsetzung mit konzentrierter Schwefelsäure anstelle eines undefinierbaren Teers ein wohldefiniertes Reaktionsprodukt entsteht.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Versetzen Sie in einem Reagenzglas ca. 2 mL konzentrierte Ameisensäure mit ca. 1 mL konzentrierter Schwefelsäure. Sollte es bei Raumtemperatur noch nicht zu einer Gasentwicklung kommen, erwärmen Sie ein wenig.
Welches Gas entsteht bei der Reaktion? Formulieren Sie die Reaktionsgleichung. Geben Sie die Lewisformeln von Ameisensäure und des entstehenden Gases an.
Verdünnte Schwefelsäure löst viele Metalle wie Eisen, Aluminium und Zink unter Wasserstoffentwicklung zu Sulfaten auf.
Man übergieße in einem Reagenzglas Zinkgranalien mit verdünnter Schwefelsäure, der man einige Tropfen konzentrierter Schwefelsäure beimischt. Das Zink wird aufgelöst, und Wasserstoff entweicht.
Formulieren Sie die Reaktionsgleichung. Welche Gefahr geht von dieser Reaktion aus?
Konzentrierte Schwefelsäure verhält sich zum Beispiel gegenüber Eisen ganz anders als die verdünnte Säure. Sie löst das Metall bei Zimmertemperatur nicht auf. Bei höherer Temperatur bildet sich das Sulfat, aber es wird kein Wasserstoff frei, sondern es entwickelt sich Schwefeldioxid, SO2.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie in einem Reagenzglas zu einigen Eisenspänen (Eisenpulver) ca. 3 mL konzentrierte Schwefelsäure und erwärmen Sie das Gemisch in der Bunsenbrennerflamme bis zur einsetzenden Gasentwicklung. Halten Sie in das entweichende Gas einen feuchten Universalindikatorstreifen.
Formulieren Sie die Reaktionsgleichung. Erklären Sie die Farbänderung des Indikatorpapiers. Welche Gefahr geht von dieser Reaktion aus?
Ein unmittelbarer Nachweis für das Vorhandensein von Schwefel in der Schwefelsäure: im oberen Teil des Reagenzglases bildet sich ein gelber Beschlag von festem Schwefel, und gelbe Schwefeltröpfchen scheiden sich ab. Entweichendes Schwefeldioxid, und manchmal auch Hydrogensulfid, sind am Geruch zu erkennen.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie eine Zinkgranalie in ein trockenes Reagenzglas (keine Späne oder gar Pulver verwenden – beides reagiert zu heftig!). Geben Sie ca. 3 mL konzentrierte Schwefelsäure hinzu und erwärmen Sie die Mischung, bis eine merkliche Umsetzung unter Gasentwicklung einsetzt. Im oberen Teil des Reagenzglases bildet sich ein gelber Beschlag und gelbe Tröpfchen scheiden sich ab.
Formulieren Sie die Reaktionsgleichungen, jeweils eine für jedes entstehende Produkt. Welche Gefahr geht von dieser Reaktion aus?
Warum ist es unkorrekt, Schwefel, H2S und SO2 alle in eine Gleichung hinein zu schreiben, so im Stil: 8 Zn + 3 H2SO4 + 16 H+ → 8 Zn2+ + S + H2S + SO2 + 10 H2O? Geht doch!
Verdünnte und konzentrierte Schwefelsäure reagieren völlig verschieden. Woran mag das liegen?
Der hohen Reaktivität nicht-protolysierter H2SO4-Moleküle steht eine geringe Reaktivität von Sulfat-Ionen, SO42−, gegenüber. Geringe Reaktivität bedeutet oft auch hohe Bildungstendenz – ein Zusammenhang, der sich bei den Reaktionen von Lösungen der schwefligen Säure zeigt. Deren typische Eigenschaft ist ihr Reduktionsvermögen bei Reaktionen, in deren Verlauf die schweflige Säure in Sulfat übergeht. Die im folgenden Versuch hergestellte schweflige Säure ist eine unbeständige, nur in wässriger Lösung existierende, schwache Säure, die beim Lösen von Schwefeldioxid in Wasser entsteht; ihre Salze heißen Sulfite und Hydrogensulfite.
Stellen Sie zunächst eine Lösung von Natriumhydrogensulfit her, indem Sie etwa eine Spatelspitze des Salzes in 1–2 mL Wasser lösen. Geben Sie dann in ein zweites Reagenzglas ebenfalls 1–2 mL Wasser und setzen Sie einen Tropfen Kaliumiodid-Iod-Lösung hinzu. Schütten Sie dann die erste zur zweiten Lösung.
Deuten Sie Ihre Beobachtungen durch Reaktionsgleichungen.
Konzentrierte Salpetersäure ist ähnlich aggressiv wie konzentrierte Schwefelsäure. In sehr verdünnter Form ist sie dagegen ein natürlich vorkommender Stoff: in Blitzen reagieren die Luftbestandteile Stickstoff und Sauerstoff zu „Stickoxiden“; zusammen mit Regenwasser bildet sich dann Salpetersäure.
Salpetersäure ist in mehreren Konzentrationen laborüblich. Rauchende Salpetersäure ist ca. 95%ig; sie ist durch Stickstoffdioxid gelb bis rotbraun gefärbt und gibt an der Luft Stickstoffdioxid-Dämpfe ab (daher auch „rote rauchende Salpetersäure“ genannt). Konzentrierte Salpetersäure ist ca. 69%ig, verdünnte Salpetersäure etwa 12%ig. Salpetersäure ist ein kräftiges Oxidationsmittel. Besonders die konzentrierten Lösungen sind sehr aggressiv. Ähnlich wie bei Schwefelsäure hängt auch das Verhalten von Salpetersäure gegenüber Metallen erheblich von der Säurekonzentration ab.
Der Umgang mit Salpetersäure (ab 5 %) wird durch eine Betriebsanweisung geregelt.
Achten Sie beim Fortschreiten dieser Reaktion besonders auf deren schnell zunehmende Geschwindigkeit – ein wesentlicher Aspekt, wenn entgegen der Betriebsanweisung einmal viel größere Mengen der Reaktionspartner zusammenfinden.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
In ein Reagenzglas gebe man zu 1–2 mL konzentrierter Salpetersäure 1–2 Zinkgranalien (keine Späne oder gar Pulver verwenden – beides reagiert zu heftig!). Es tritt heftige Entwicklung von rotbraunem Stickstoffdioxid-Gas auf. Nachdem man dies beobachtet hat, wird die Reaktion durch Verdünnen mit viel Wasser beendet.
Stellen Sie die Reaktionsgleichung auf. Worin besteht die Gefahr? Warum wird die Reaktion immer heftiger? Was müsste man tun, um 1 kg Zink sicher zu Zinknitrat umzusetzen?
Wieviel Kilogramm Zinknitrat-Dihydrat lassen sich aus 1 kg Zink durch die Reaktion mit konzentrierter Salpetersäure herstellen?
Eine auf den ersten Blick erstaunliche Reaktion: das Metall Zinn reagiert nicht zu Zinn-nitrat, sondern zu Zinndioxid, SnO2, das als Mineral Cassiterit („Zinnstein“) seit dem Beginn der Bronzezeit als Rohstoff für die namengebende Kupfer-Zinn-Legierung begehrt ist.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
In einem Reagenzglas wird etwas Zinnfolie oder eine Zinngranalie (kein Zinnpulver verwenden – es reagiert viel zu heftig!) mit konzentrierter Salpetersäure unter Bewegen des Glases mäßig erwärmt. Das Zinn wird dabei zu weißem Zinndioxid oxidiert, das ungelöst bleibt. Bei der Reaktion entstehen rotbraune Dämpfe von Stickstoffdioxid.
Stellen Sie die Reaktionsgleichung auf. Worin besteht die Gefahr? Haben Sie schon eine erste Idee, warum kein Zinn-nitrat entsteht? (In der Grundvorlesung wird ausführlich darüber geredet; es geht darum, was alles den Unterschied zwischen Metallen und Nichtmetallen ausmacht.)
Wieviel Kilogramm einer Bronze aus 15 Masse-% Zinn und 85 Masse-% Kupfer lässt sich aus 1 kg Zinndioxid herstellen?
Eine der Reaktionen, bei denen reichlich farbbloses NO gebildet wird, so dass Sie dessen spontane Oxidation zu braunem NO2 beim Kontakt mit Luft sehr schön beobachten können.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Man bereite in einem Reagenzglas durch Versetzen von etwas konzentrierter Salpetersäure mit etwas mehr als dem gleichen Volumen Wasser halbkonzentrierte Salpetersäure, gebe einige Zinkgranalien zu und erwärme. Anders als bei der Reaktion mit konzentrierter Säure entwickelt sich ein nur schwach braunes Gas: es entsteht ein Gemisch von viel farblosem Stickstoffmonoxid mit etwas braunem Stickstoffdioxid.
Stellen Sie die Reaktionsgleichung für die Bildung von NO auf. Worin besteht die Gefahr? Zur Wiederholung noch einmal: Warum wäre eine Reaktionsgleichung nicht korrekt, bei der auf der Seite der Produkte so etwas wie NO + NO2 stünde? Es entsteht doch wirklich nicht nur NO!
Noch eine Frage, die erst später ausführlich aufgegriffen wird: es ist schon sehr erstaunlich, dass viele Gase, die mit Sauerstoff ein stabiles Reaktionsprodukt bilden, dies bei Raumtemperatur aber ebensowenig tun wie feste (Holz, Magnesium) oder flüssige (Benzin) Brennstoffe – in allen diesen Fällen muss gezündet werden. Beispiele für solche Gase sind Wasserstoff, Methan (Erdgas) oder Schwefeldioxid. Bei NO jedoch ist die Reaktion auch bei Raumtemperatur offensichtlich völlig ungehemmt. Haben Sie eine erste Idee, womit das zu tun haben könnte?
Salzsäure, Schwefelsäure, Salpetersäure: jede dieser Säuren hat eine eigene Chemie – solange sie nicht verdünnt vorliegen. Vergleichen Sie selbst:
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
In einem Reagenzglas verdünne man etwas verdünnte Salpetersäure auf das doppelte Volumen, setzte einige Zinkgranalien zu und erwärme. Es entwickelt sich ein farbloses Gas, das sich auch bei Luftzutritt an der Mündung des Reagenzglases nicht braun färbt: mit der verdünnten Säure entsteht Wasserstoff.
Bringen Sie es noch einmal auf den Punkt: warum verlieren die Säuren beim Verdünnnen ihre Individualität?
Wieviel Kilogramm des als Beiz- und Bleichmittels verwendeten Zinknitrat-Hexahydrats lässt sich aus 1 kg Zink durch die Reaktion mit verdünnter Salpetersäure herstellen?
Eine ungewöhnliche Reaktion: viele Versuche haben bisher gezeigt, dass selbst bei hoher Reaktivität einer Säure deren Säureanion kaum reaktiv ist. Schwefelsäure und Sulfat sind Beispiele und auch Salpetersäure und Nitrat zeigen im Großen und Ganzen dieses Prinzip. Mit dem starken Reduktionsmittel Zink lässt sich jedoch Nitrat sogar in alkalischer Lösung reduzieren.
In einem Reagenzglas wird eine Spatelspitze Zinkstaub mit etwa 6 Tropfen verdünnter Salpetersäure übergossen. Sogleich fügt man dazu 2 mL Natronlauge und erhitze zum Sieden. In die Dämpfe wird ein Streifen feuchtes Indikatorpapier so gehalten, dass es die Wände nicht berührt. Es färbt sich bald durch die Einwirkung von Ammoniak-Gas, das auch durch seinen charakteristischen Geruch nachweisbar ist.
Formulieren Sie die Reaktionsgleichung. Warum müssen sie darauf achten, dass das pH-Papier nicht das Reagenzglas berührt?
Umsetzungen, deren Gefahr in der unbeabsichtigten Freisetzung nitroser Gase besteht, lassen sich so steuern, dass eine besonders wichtige Verbindung gezielt und rein hergestellt werden kann: Stickstoffmonoxid, NO, ein grosstechnisches Zwischenprodukt auf dem Wege von Ammoniak zu Salpetersäure, und zugleich ein Hormon, das zur Erweiterung der Blutgefäße führt.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie in den Rundkolben der für diesen Versuch zur Verfügung gestellten Gasentwicklungsapparatur eine Spatelspitze Kupferspäne und versetzen Sie diese mit einer Lösung aus zwei Teilen Wasser und einem Teil konzentrierter Salpetersäure. Nachdem die Luft im Reaktionsgefäß verdrängt ist, stülpe man über die Öffnung des Gasentbindungsrohres ein mit Wasser gefülltes Reagenzglas. Dabei beobachtet man im Kolben mehr oder weniger rotbraune Dämpfe, die ein Gemisch von Stickstoffmonoxid und Stickstoffdioxid darstellen. Beim Durchgang durch das Wasser reagiert Stickstoffdioxid. Das im Reagenzglas aufgefangene Gas besteht nur aus farblosem Stickstoffmonoxid. Hebt man das Reagenzglas aus dem Wasser heraus, so färbt sich der Inhalt von der Mündung her schnell braun, weil sich das Stickstoffmonoxid mit dem Luftsauerstoff zu Stickstoffdioxid umsetzt.
Formulieren Sie hier nicht nur die Reaktionsgleichung für die Umsetzung des Kupfers mit der Säure, sondern auch die Gleichung für die Reaktion des Stickstoffdioxids mit Wasser und für die Reaktion des Stickstoffmonoxids mit Luftsauerstoff.
Die Grundchemikalie Ammoniak wird großtechnisch aus den Elementen über das Haber-Bosch-Verfahren gewonnen. In Form von Harnstoff und Ammoniumsalzen findet Ammoniak ausgedehnte Verwendung als Dünger. Als Ausgangsstoff für die Salpetersäureherstellung (Ostwald-Verfahren) erschließt Ammoniak den Einstieg in die technische Stickstoffchemie.
Die verdünnte Ammoniaklösung des Labors ist ca. 10%ig, konzentrierte Ammoniaklösung ist ca. 25%ig; die konzentrierte Lösung hat eine Dichte von 0,91 g cm−3. Eine aufgrund der hohen Entweichungstendenz von Ammoniakgas kaum handhabbare gesättigte Lösung in Wasser ist mehr als 30%ig (Raumtemperatur).
Wieviel molar ist 25%ige Ammoniaklösung?
Der Umgang mit Ammoniaklösung wird durch eine Betriebsanweisung geregelt. Die dort als gefährlich aufgeführten Umsetzungen sind zum Teil so brisant, dass Sie diese – wie zum Beispiel die Umsetzung mit Iod zu Triiodnitrid („Iodstickstoff“, NI3) – nur in der Grundvorlesung kennenlernen werden, nicht aber in Praktikumsversuchen.
Einige der wichtigsten Konzepte der Chemie sind dem Ausgleich eines unterschiedlichen Angebots an negativer Ladungsdichte gewidmet. Ammoniak ist ein Lehrbuchbeispiel für ein Molekül, dessen Reaktionen durch die Anwesenheit eines freien Elektronenpaars am Stickstoffatom bestimmt sind. Stoffe, deren Reaktivität auf verfügbaren Elektronenpaaren beruht, sind in Säure-Base-Konzepten die Base, in Elektrophilie-Nukleophilie-Konzepten das Nukleophil und schließlich, als Spezialfall einer Säure-Base-Betrachtung, können sie gegenüber Metall-Ionen als Liganden auftreten.
Versetzen Sie etwa 2 mL destilliertes Wasser mit 2 Tropfen des Indikators Phenolphthalein. Geben Sie nun 1–2 Tropfen 6 m Ammoniaklösung zu. Versetzen Sie die jetzt rote Lösung mit einigen mL einer konzentrierten NH4Cl-Lösung, bis die rote Farbe wieder verschwindet.
Erklären Sie Ihre Beobachtung.
Stellen Sie eine Kupfersulfatlösung her, indem Sie ca. 2 Spatelspitzen Kupfer(II)-sulfat in etwa 2 mL destilliertem Wasser lösen. Tropfen Sie nun langsam 6 m Ammoniaklösung zu. Es bildet sich zunächst ein Niederschlag, der sich bei weiterer Zugabe von Ammoniaklösung allmählich wieder auflöst und eine tiefblaue Lösung entsteht.
Deuten Sie Ihre Beobachtungen durch Reaktionsgleichungen. Woher kommt die hellblaue Farbe der reinen Kupfersulfatlösung?
Neben der grosstechnisch bedeutsamen Oxidation von Ammoniakgas zu NO im Ostwald-Verfahren lässt sich eine wissenschaftshistorisch bedeutende Oxidation von Stickstoff(−III) unter milden Bedingungen in wässriger Lösung beobachten: die Symproportionierung von Ammonium- und Nitrit-Stickstoff zu elementarem Stickstoff, N2.
Jeweils 1 Spatel Ammoniumchlorid und Natrium- oder Kaliumnitrit werden in wenig Wasser gelöst. Es wird vorsichtig erwärmt, bis Gasentwicklung einsetzt. Ein glimmender Holzspan erlischt, wenn er in den Gasraum des Reagensglases eingetaucht wird.
Formulieren Sie Reaktionsgleichung (für Nitrit auch die Lewis-Formel) für die Bildung von Stickstoff aus einem Ammoniumsalz und einem Nitrit.
Die historische Bedeutung des Versuchs ist die folgende: Der gebildete Reinstickstoff weist eine geringfügig kleinere Dichte auf als Luft, der durch chemische Reaktionen Wasser (durch Trockenmittel entfernt), Kohlendioxid (durch Base entfernt) und Sauerstoff (durch Reduktionsmittel entfernt) entzogen wird. Können Sie dies erklären und überblicken Sie, was in der Folge entdeckt wurde?
„Natronlauge“ ist kein systematischer Name, sondern eine althergebrachte Bezeichnung für eine wässrige Lösung von Natriumhydroxid, NaOH, in Wasser. Dasselbe gilt für Kalilauge. Vor allem die konzentrierten Laugen sind deutlich gefährlicher als etwa eine gleichkonzentrierte Salzsäure, da sie viel hartnäckiger an der Haut haften als eine HCl-Lösung und nur schwer durch Wasser abgespült werden. Entsprechend gefährlich ist das heute vor allem auf historischen Jahrmärkten wieder in Mode gekommene „Seifensieden“ in offenen Bottichen: Mit den seit langem verfügbaren Chemikalien Branntkalk (Calciumoxid) oder Löschkalk (Calciumhydroxid) wird „Sodalösung“ (Natriumcarbonatlösung) „kaustifiziert“ (von lat. causticus ätzend, beizend), anschließend wird die entstandene Lauge mit Fett umgesetzt, wobei dieses in Glycerin und Seife übergeht. Eine weitere Anwendung von Natronlauge im Alltag: 3–5%ige Lauge wird bei der Herstellung von Laugenbrezen verwendet.
Haben Sie eine Idee, welche Chemie hinter dem Kaustifizieren und dem Seifensieden steckt?
Beim Auflösen von 40 g der handelsüblichen NaOH-Plätzchen in Wasser zu 1 L wird eine 1-molare Lösung erhalten. Beim Auflösen von 56 g KOH-Plätzchen zu 1 L wird jedoch eine nur 0,85-molare Lösung erhalten. Woran liegt das?
Verdünnte Natronlauge ist 7–8%ig (2 m), konzentrierte Natronlauge enthält 40 % Natriumhydroxid.
Der Umgang mit Alkalilaugen wird durch eine Betriebsanweisung geregelt.
Die Betriebsanweisung zu Alkalilaugen führt deren Umsetzung mit Metallen als besondere Gefahr auf. Der Versuch soll aufzeigen, wieso.
Geben Sie eine Spatelspitze Aluminium-Grieß in ein Reagenzglas und stellen Sie dieses (vorsichtshalber) in ein hohes Becherglas. Geben Sie nun ein paar Tropfen verdünnte Natronlauge hinzu. Die Reaktion benötigt einen kurzen Augenblick bis sie in Gang kommt, verläuft dann aber äußert heftig (Aufschäumen des Reaktionsgemisches). Fangen Sie das entstehende Gas mit einem zweiten Reagenzglas auf und führen Sie eine Knallgasprobe durch.
Um welches Gas handelt es sich? Formulieren Sie die Reaktionsgleichung. Warum ist die Umsetzung mit Aluminium eine Gefahr?
Die Betriebsanweisungen zu Alkalilaugen und festen Alkalihydroxiden führt deren Umsetzung mit unedlen Metallen als besondere Gefahr auf. Der Versuch soll aufzeigen, wieso.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
In einem Reagenzglas werden zwei NaOH-Plätzchen mit zwei Spatelspitzen Zinkpulver gemischt. Das Reagenzglas wird mit einem durchbohrten Gummistopfen verschlossen, in dessen Öffnung sich eine eingesteckte Pasteur-Pipette befindet. Die Mischung wird nun unter ständigem Bewegen des Reagenzglases über dem Bunsenbrenner erhitzt. Wenn eine deutliche Gasentwicklung zu erkennen ist, wird das an der Pipetten-Öffnung austretende Gas mit dem Bunsenbrenner entzündet.
Um welches Gas handelt es sich? Formulieren Sie die Reaktionsgleichung. Warum ist die Umsetzung mit Zink eine Gefahr?
Die Betriebsanweisung zu Alkalilaugen führt deren Umsetzung mit Ammoniumsalzen als besondere Gefahr auf. Der Versuch soll aufzeigen, wieso.
Geben Sie etwa vier Spatelspitzen Ammoniumchlorid in ein Reagenzglas und lösen Sie das Salz in ca. 2 mL destilliertem Wasser. Versetzen die Lösung mit ein paar Tropfen Natronlauge (6 m). Halten Sie anschließend ein angefeuchtetes pH-Papier an die Öffnung des Reagenzglases (ohne dieses mit dem Papier zu berühren).
Welches Gas können Sie so nachweisen (Reaktionsgleichung)? Warum führt die Betriebsanweisung die Reaktion mit Ammoniumsalzen als Gefahr auf?
Noch einmal, nur zur Wiederholung: Warum müssen sie darauf achten, das pH-Papier nicht das Reagenzglas berühren zu lassen?
Versetzt man wässrige Lösungen von Übergangsmetallsalzen mit Natron- oder Kalilauge, beobachtet man zumeist die Bildung eines Niederschlags, es bildet sich ein Metallhydroxid. Einige dieser Hydroxide lassen sich nicht nur durch Zugabe von Säure wieder in Lösung bringen, sondern auch durch einen Überschuss an Lauge. Stoffe (im engeren Sinn Hydroxide), die sowohl als Säure als auch als Base reagieren können, bezeichnet man als amphoter.
Lösen Sie etwa zwei Spatelspitzen Aluminiumchlorid in 1–2 mL destilliertem Wasser und tropfen Sie langsam 6 m Natronlauge hinzu. Es bildet sich zunächst ein Niederschlag, der sich durch langsame Zugabe von einem Überschuss Natronlauge wieder auflöst. Geben Sie zu dieser klaren Lösung nun tropfenweise und unter gutem Durchmischen langsam 6 m Salzsäure, bis sich wieder ein Niederschlag bildet. Bei Zugabe von weiterer Säure löst sich dieser wieder auf.
Deuten Sie Ihre Beobachtungen durch Reaktionsgleichungen.
Wasserstoffperoxid-Lösung ist eine farblose Flüssigkeit, in der die gegen Zerfall in Wasser und Sauerstoff metastabile, blassblaue Flüssigkeit Wasserstoffperoxid in verdünnter Form vorliegt. Wasserstoffperoxid ist eine schwache Säure und gegenüber den meisten Stoffen ein starkes Oxidationsmittel, worauf die Verwendung als Bleich- und Desinfektionsmittel beruht. In hochkonzentrierter Form ist es sowohl als Einzel- als auch als Komponentenraketentreibstoff einsetzbar. Lösungen in Wasser sind in mehreren Konzentrationen handelsüblich. Im Labor wird meist eine 30%ige Lösung verwendet, während im Alltag ca. 10%ige Lösung (zum Blondieren von Haaren) oder ca. 3%ige Lösung („Wasserstoffsuperoxid“, zur Desinfektion) eingesetzt wird.
Haben Sie eine Idee, wieso Wasserstoffperoxid, zum Beispiel bei der Papierherstellung (Zellstoffbleiche), als ein ökologisch günstig zu bewertendes („grünes“) Bleichmittel eingestuft wird?
In der Ausgabe vom 23. August 2008 berichtet die Süddeutsche im Münchner Teil:
Ätzende Chemikalie löst Feueralarm aus: In einem Versicherungsgebäude an der Dieselstraße ist am Donnerstagmittag das ätzende Gas Wasserstoffperoxid ausgetreten. Im zweiten Untergeschoss des Hauses entwich das Gas aus einem 30-Liter-Kunststoffbehälter, der die Chemikalie Bamacid enthielt. Diese wird zur Säuberung von Brauch- und Abwässern verwendet. [Anm.: Bamacid ist ein Entkeimungsmittel, das Wasserstoffperoxid und ein Wasserstoffperoxid-Derivat enthält.] Nachdem das Behältnis morgens gewechselt worden war, kam es aufgrund von Verunreinigungen zu einem Temperaturanstieg. Dadurch entwickelte sich ein Überdruck, der den Deckel absprengte. …
Können Sie den Hergang erklären? Wasserstoffperoxid ein Gas? Temperaturanstieg durch Verunreinigungen?
Der Umgang mit Wasserstoffperoxid-Lösung wird durch eine Betriebsanweisung geregelt.
Starke Säuren wie Schwefelsäure können das Anion von Peroxiden protonieren, wodurch Wasserstoffperoxid erhalten wird.
Führen Sie diesen Versuch in Zweiergruppen aus.
Geben Sie in ein Becherglas oder Erlenmeyerkolben etwa 10 ml eiskalte 20%ige Schwefelsäure und fügen Sie in kleinen Mengen ca. 2 g Bariumperoxid hinzu. Um die Reaktion gut zu kühlen, geben sie während der Zugabe des Peroxids immer wieder kleine Eisstückchen dazu. Während der Reaktion fällt schwerlösliches Bariumsulfat aus. Nach der vollständigen Zugabes des Bariumperoxides wird mit festem Bariumcarbonat versetzt, damit die überschüssige Säure abreagiert. Filtrieren Sie anschließend das Reaktionsgemisch und benutzen Sie die so erhaltene Wasserstoffperoxid-Lösung in den folgenden Versuchen.
Formulieren Sie die Reaktionsgleichungen.
Bringen Sie ca. 2 mL der im vorherigen Versuch hergestellten Wasserstoffperoxid-Lösung mit Natronlauge auf einen pH-Wert des stark basischen Bereiches und fügen Sie einige Tropfen einer MnSO4-Lösung zu.
Was können Sie beobachten?
Wasserstoffperoxid kann als Quelle zur Synthese kleiner Mengen reinen Sauerstoffs genutzt werden.
Geben Sie eine Spatelspitze Braunstein, MnO2, in ein Reagenzglas und tropfen Sie vorsichtig ca. 0,5 mL der zuvor hergestellten Wasserstoffperoxid-Lösung hinzu. Es kommt zu einer Gasentwicklung. Führen Sie eine Glimmspannprobe durch, um einen Hinweis auf die Identität des Gases zu bekommen.
Welches Gas haben Sie nachgewiesen? Welche Funktion hat MnO2 bei dieser Reaktion? Formulieren Sie die Reaktionsgleichung.
-->Im Mittelpunkt des Vorkurses stehen zwei handwerkliche Aspekte: Sicheres Arbeiten stand natürlich am Anfang, jetzt kommt genaues Arbeiten hinzu – Sie lernen die „Maßanalyse“ kennen, an der Sie nur Freude haben werden, wenn Sie das Abmessen von Volumina und das Wiegen ausreichend genau betreiben.
Die Maßanalyse beruht auf der präzisen Bestimmung von Flüssigkeitsvolumina. Die hierbei verwendeten Volumenmessgeräte sind Messzylinder, Messkolben, Pipetten und Büretten. Der Inhalt der Messgefäße wird in mL angegeben und ist auf eine Temperatur von 20 °C geeicht. Messkolben und Messzylinder sind auf Einguss (Kennzeichnung „In“) geeicht, das heißt, dass sie bei der entsprechenden Eichtemperatur das angegebene Volumen fassen. Büretten und Pipetten sind dagegen in der Regel auf Auslauf (Kennzeichnung „Ex“) geeicht, das heißt, dass beim Entleeren der auf dem Messgefäß angegebene Inhalt abgegeben wird. Geeichte Messgefäße werden (nach der Deutschen Eichordnung) in Geräte der Klassen A und B unterteilt, wobei die Geräte der Klasse B eine doppelt so große Fehlergrenze besitzen. Um welche Klasse es sich handelt, erkennen Sie an der Kennzeichnung „A“ bzw. „B“ auf dem jeweiligen Messgefäß.
Messkolben sind Standkolben mit einem langen, engen Hals, auf dem sich eine Eichmarke befindet. Sie dienen vor allem zur Herstellung von Maßlösungen. Befüllen Sie dazu den Messkolben zunächst mit einer genau abgewogenen Substanzmenge oder mit einem Standardkonzentrat und füllen Sie den Kolben mit destilliertem Wasser bis zur Hälfte auf. Lösen oder durchmischen Sie nun die Substanz durch vorsichtiges Schwenken. Um das Volumen so exakt wie möglich abzumessen, befüllen Sie den Kolben nun zunächst bis etwa ein oder zwei Zentimeter unterhalb der Eichmarke. Warten Sie dann 1–2 Minuten, damit gegebenenfalls Flüssigkeitsreste von der Glaswand ablaufen können. Verwenden Sie dann eine Einwegpipette und geben Sie gerade so viel Flüssigkeit hinzu, bis der unterste Punkt des Flüssigkeitsspiegels (der „Meniskus“) mit der Eichmarke zusammenfällt. Die Markierung muss beim Ablesen auf Augenhöhe liegen, so dass der vordere Teil des Ringes den hinteren genau überdeckt. Verschließen Sie anschließend den Messkolben und schütteln Sie ihn über Kopf, um die Lösung gut zu durchmischen.
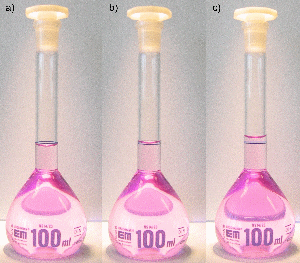
Befüllen eines Messkolbens: a) zu wenig, b) richtig befüllt, c) viel zu viel.
Messzylinder sind Standzylinder, auf denen sich eine Graduierung befindet, mit deren Hilfe sich ein bestimmtes Volumen abmessen lässt. Messzylinder sind weniger genau als Messkolben und sollten daher nur zur Grobmessung verwendet werden. (Unter keinen Umständen sollten Sie Bechergläser oder Erlenmeyerkolben zur Abmessung von Flüssigkeitsmengen zur Analyse verwenden, da die Volumenmarkierung bei diesen Geräten nur als grober Anhaltspunkt angebracht sind.) Achten Sie beim Abmessen auch hier darauf, dass die Markierung für das gewünschte Volumen mit dem untersten Punkt des Flüssigkeitsspiegels zusammenfällt und dass Sie diese Markierung in Augenhöhe betrachten.
Pipetten lassen sich in Voll- und Messpipetten unterteilen. Vollpipetten sind Glasröhren mit einer Ausbuchtung in der Mitte. Ihr Inhalt wird durch eine Eichmarke am oberen Ende markiert. Messpipetten sind Glasröhren, die im Unterschied zu den Vollpipetten eine Graduierung besitzen.
Zur Abmessung eines gewünschten Volumens tauchen Sie die Spitze der Pipette in die entsprechende Flüssigkeit und ziehen diese bis über die Eichmarke auf. Legen Sie die Pipettenspitze an der Glaswand des schräg gehaltenen Gefäßes an und lassen Sie so viel Flüssigkeit herauslaufen, bis der Meniskus auf der Eichmarke liegt. Wichtig ist, dass Sie die Pipette dabei gerade halten und die Eichmarke auf Augenhöhe liegt. Streifen Sie nun die Pipettenspitze an der Glaswand ab und entleeren Sie die Flüssigkeit in das vorgesehene Gefäß. Dabei liegt die Pipettenspitze wieder an der Glaswand des schräg gehaltenen Gefäßes an. Nachdem die Pipette abgelaufen ist, warten Sie noch solange wie für den Pipettentyp vorgesehen (siehe unten), bevor Sie die Pipette nach oben hin von der Gefäßwand abziehen. Da Pipetten auf Auslauf geeicht sind, wurde nun der auf der Pipette angegebenen Inhalt abgegeben. Dass in der Pipettenspitze ein Rest zurückbleibt, ist bei der Eichung auf Auslauf berücksichtigt, so dass dieser nicht, zum Beispiel durch Ausblasen, zusätzlich entleert werden darf.
Als Pipettierhilfe zum Aufsaugen und Ablassen der Flüssigkeit wird ein Peleusball verwendet. Dieser besitzt drei Ventile. Ventil A dient zum Ausdrücken der Luft aus dem Ball, Ventil S zum Ansaugen der Flüssigkeit und Ventil E zum Entleeren der Pipette. Der Peleusball wird auf die Pipette gesetzt, Ventil A durch Zusammendrücken mit Daumen und Zeigefinger geöffnet und die Luft aus dem Gummiball gedrückt. Durch Druck auf Ventil S kann nun die Flüssigkeit aufgezogen und durch Druck auf Ventil E wieder abgelassen werden.

Peleusball
Mess- und Vollpipetten werden in unterschiedliche Genauigkeitsklassen eingeteilt. Bei Pipetten der Klasse A ist die Ablaufzeit durch eine verengte Spitze verlängert, so dass bereits während des Ablaufens die Flüssigkeit von der Glaswand nachläuft. Die Pipetten der Klasse B besitzen eine erheblich kürzere Ablaufzeit, dafür aber auch eine doppelt so große Fehlergrenze. Neben den Typen A und B gibt es noch Pipetten der Klasse AS. Es handelt sich dabei um Pipetten mit der gleichen Fehlergrenze wie jene der Klasse A, aber mit deutlich kürzeren Ablaufzeiten. Jedoch benötigen diese nach dem Ablauf der Flüssigkeit eine kurze Wartezeit, meist 15 Sekunden. Die genaue Wartezeit finden Sie auf der jeweiligen Pipette des Typs AS, z.B. „Ex+15s“ für eine Wartezeit von 15 Sekunden nach dem Ablauf der Flüssigkeit. Pipetten der Klassen A und B benötigen keine Wartezeit. Zur Übersicht finden Sie die Fehlergrenzen und Ablaufzeiten ausgewählter Vollpipetten in der folgenden Tabelle.
| Klasse A | Klasse AS | Klasse B | ||||
|---|---|---|---|---|---|---|
| Nennvolumen [mL] | max. Fehler [%] | Ablaufzeit [s] | max. Fehler [%] | Ablaufzeit [s] | max. Fehler [%] | Ablaufzeit [s] |
| 5 | ± 0,30 | 15–30 | ± 0,30 | 7–11 | ± 0,60 | 7–30 |
| 10 | ± 0,20 | 15–40 | ± 0,20 | 8–12 | ± 0,40 | 8–40 |
| 20 | ± 0,15 | 25–50 | ± 0,15 | 9–13 | ± 0,30 | 9–50 |
| 25 | ± 0,13 | 25–50 | ± 0,13 | 10–15 | ± 0,26 | 10–50 |
| 50 | ± 0,10 | 30–60 | ± 0,10 | 13–18 | ± 0,20 | 13–60 |
| 100 | ± 0,08 | 40–60 | ± 0,08 | 25–30 | ± 0,16 | 25–60 |
Beachten Sie, dass im Praktikum möglichst genaues Arbeiten geübt werden soll. Verwenden Sie daher Pipetten der Typen A oder AS und beachten Sie die angegebene Wartezeit.
Büretten sind graduierte Glasrohre mit einem Hahn am unteren Ende zur regelbaren Abgabe genau bekannter Flüssigkeitsmengen. Zur Benutzung der Bürette spannen Sie diese senkrecht in ein Stativ ein und befüllen sie mit Hilfe eines Trichters zunächst mit ein paar Millilitern der Flüssigkeit. Öffnen sie vorsichtig den Hahn, bis die Flüssigkeit herauszulaufen beginnt und schließen Sie den Hahn dann wieder. Die Ablaufspitze sollte jetzt vollständig mit Flüssigkeit gefüllt sein, es darf sich unter keinen Umständen noch Luft im unteren Teil der Bürette befinden. Füllen Sie die Bürette nun mit Flüssigkeit bis ca. 0,5 cm oberhalb der Nullmarke auf und lassen Sie durch vorsichtiges Öffnen des Hahns die Flüssigkeit bis zur Nullmarke ab. Wie bei den anderen Messgefäßen wird auch hier der Flüssigkeitsstand an der tiefsten Stelle des Meniskus abgelesen. Das Ablesen wird dabei durch einen farbigen Streifen (Schellbachstreifen) auf der Rückwand der Bürette erleichtert. Auf der Höhe des Flüssigkeitsspiegels ist dieser Streifen fast vollständig eingeschnürt. Eine Ausnahme bilden intensiv farbige Flüssigkeiten, bei denen an der oberen Kante ablesen wird.
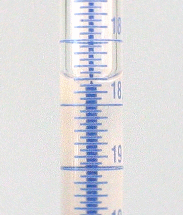
Schellbachstreifen
Sind Sie am Endpunkt einer Titration angelangt, wird sich der Meniskus in den seltensten Fällen genau auf einem Teilstrich der Graduierung befinden. Orientieren Sie sich in einem solchen Fall an den Markierungen ober- und unterhalb des Meniskus und schätzen Sie den Wert dazwischen so genau wie eben möglich auf die zweite Nachkommastelle ab. Verwenden Sie beispielsweise eine 25-mL-Bürette mit Teilstrichen von 0,05 mL, und der Meniskus liegt zwischen 11,15 und 11,20 mL, so schätzen Sie ab, ob er bei 11,16, 11,17, 11,18 oder 11,19 mL liegt.
Wird eine Bürette mit Glashahn benutzt, so muss dieser vor der Benutzung leicht gefettet werden (dies entfällt bei einem Teflonhahn). Achten Sie darauf, dass kein Schlifffett in die Hahnbohrung gelangt. Der Schliff sollte gleichmäßig klar erscheinen und der Hahn leicht drehbar sein. Verwenden Sie zum Fetten nur das dafür geeignete Schlifffett.
Nehmen Sie einen 250-mL-Messkolben zur Hand und entleeren Sie in diesen fünfmal eine 50 mL Vollpipette mit destilliertem Wasser.
Konnten Sie den Messkolben exakt befüllen? Falls nicht, überlegen Sie, wieso Sie die Eichmarke nicht genau getroffen haben.
Befüllen Sie einen 250-mL-Messkolben mit gekühltem Wasser und beobachten Sie was passiert, wenn das Wasser Raumtemperatur erreicht hat.
Erklären Sie Ihre Beobachtung.
Wie wichtig eine gründliche Reinigung von Laborgeräten ist, haben Sie bereits in einem früheren Versuch gelernt. Dies gilt besonders für die zur quantitativen Analyse verwendeten Messgefäße, die sowohl frei von chemischen Rückständen, als auch frei von Staub und Fett sein müssen. Ein Fettfilm in Büretten oder Pipetten führt zu einer geringeren Benetzung der Glaswand und infolge dessen zu einer Vergrößerung des abgegebenen Volumens.
Reinigen Sie daher ihre Glasgeräte immer unmittelbar nach dem Gebrauch, indem Sie gründlich mit destilliertem Wasser spülen. Verwenden Sie verdünnte Säuren oder Laugen nur bei gröberen Verschmutzungen, die sich sonst nicht beseitigen lassen. Konzentrierte Laugen sollten nicht verwendet werden, da diese über einen längeren Zeitraum das Glas angreifen. Spülen Sie ihre Messgefäße keinesfalls mit Aceton, Ethanol oder einem anderen organischen Lösemittel nach, da diese Fettspuren enthalten können, welche dann auf der Glaswand zurückbleiben. Auch das Verwenden von Druckluft zum schnelleren Trocknen der Messgefäße ist zu unterlassen, da hierbei Fett aus den Leitungen in die Glasgeräte gelangen kann. Vor der Verwendung Ihrer Bürette sollten Sie diese mit einigen Millilitern der Titrationslösung spülen, um eventuelle Rückstände oder Staub zu entfernen.
Zur Maßanalyse zählt die Detektion des Endpunktes einer chemischen Reaktion – hier die Detektion der vollständigen Reaktion einer Säure mit einer Base. Sie lernen in der begleitenden Vorlesung, dass die hierzu geeigneten Indikatoren (lat. indicare anzeigen) selbst Säuren und Basen sind und so Änderungen des pH-Wertes auf der Grundlage des folgenden Protolysegleichgewichts anzeigen können:
HInd + H2O ⇌ H3O+ + Ind−
Zu etwa 10 mL Wasser werden einige Tropfen Methylorange-Lösung gegeben. Die Lösung wird tropfenweise mit verdünnter Salzsäure versetzt, wobei sich die Farbe in charakteristischer Weise ändert. Nun wird bis zum Farbumschlag tropfenweise verdünnte Natronlauge zugegeben.
Zu etwa 10 mL Wasser werden einige Tropfen Phenolphthalein-Lösung gegeben. Die Lösung wird tropfenweise mit verdünnter Natronlauge versetzt, wobei sich die Farbe in charakteristischer Weise ändert. Nun wird bis zum Farbumschlag tropfenweise verdünnte Salzsäure zugegeben. Versuchen Sie eine Mischfarbe zu erreichen.
Lösungen mit genau bekanntem Gehalt (Titer) an Gelöstem heißen Maßlösungen oder eingestellte Lösungen. Die genaue Konzentration wird durch die Angabe der ungefähren Konzentration (zum Beispiel 0,1 m) und der Angabe eines in der Regel geringfügig von 1 abweichenden Faktors angegeben.
Beispiel: Eine Natronlauge der Konzentration 0,0996 m ist eine 0,1 m Maßlösung mit dem Faktor 0,996. Sie wird eingesetzt, wenn eine Vorschrift eine 0,1 m Natronlauge verlangt.
Zur Herstellung einer 0,1 m Natronlauge wiegen Sie eine genaue Menge an Natriumhydroxid (wie viel?) ab. Spülen Sie die NaOH-Plätzchen mit etwas destilliertem Wasser ab um die an der Oberfläche anhaftende Carbonatschicht zu entfernen. Lösen Sie dann das Natriumhydroxid in einem ausreichend großem Becherglas und füllen Sie die Lösung mit Hilfe eines Trichters in einen 1-L-Messkolben. Spülen Sie Reste der NaOH-Lösung im Becherglas mit etwas destilliertem Wasser in den Messkolben und füllen Sie diesen dann bis zur Eichmarkierung auf. Lagern sie Ihre Natronlauge stets gut verschlossen in einer Polyethylenflasche, damit diese möglichst kein CO2 aus der Luft aufnimmt.
Die zu bestimmende Lösung erhalten Sie in einem Messkolben (250 mL), den Sie mit destilliertem Wasser wie oben beschrieben bis zur Eichmarke auffüllen. Entnehmen Sie der Lösung nun eine genau definierte Menge mit Hilfe einer Vollpipette (in der Regel 25 oder 50 mL) und entleeren Sie diese in einen Weithals-Erlenmeyerkolben mit ausreichendem Volumen. Verdünnen Sie ihre Probelösung mit destilliertem Wasser auf etwa das doppelte Volumen und setzen Sie anschließend den für die jeweilige Titration vorgesehenen Indikator zu. Dabei reichen im Normalfall einige Tropfen der Indikatorlösung. Beachten Sie gegebenenfalls weitere Titrationsbedingungen wie zum Beispiel den pH-Wert.
Füllen Sie nun Ihre Bürette mit der entsprechenden Titratorflüssigkeit (Maßlösung) und notieren Sie den Flüssigkeitsstand (Nullmarke; muss nicht zwingend auf der 0-mL-Marke der Eichskala liegen, natürlich keinesfalls darüber). Nun können Sie mit dem Eintropfen der Maßlösung in Ihre Probelösung beginnen, wobei Sie durch andauerndes Schwenken des Erlenmeyerkolbens für eine gute Durchmischung sorgen. Zu Beginn der Titration können Sie die Maßlösung noch etwas schneller zutropfen, gegen Ende jedoch nur noch langsam, Tropfen für Tropfen. Achten Sie auf einen an der Bürettenspitze hängenden Tropfen. Ein solcher Tropfen gehört zu Ihrer verbrauchten Maßlösung. Sie können Ihn an der Glaswand des Erlenmeyerkolbens abstreifen und mit Hilfe von wenig(!) destilliertem Wasser in die Probelösung spülen.
Warten Sie nach erfolgtem Farbumschlag die Ablaufzeit der Bürette ab und lesen Sie dann den Flüssigkeitsstand ab. Es kann mitunter hilfreich sein, den Flüssigkeitsstand schon kurz vor dem vermuteten Endpunkt zu notieren, um im Falle eines Übertitrierens noch ein brauchbares Ergebnis zu erhalten. Eine weiße Unterlage, zum Beispiel ein Blatt Papier, kann die Erkennung des Umschlagspunktes erleichtern. Auch eine Vergleichslösung kann helfen, den Umschlagspunkt besser zu erkennen. Machen sie sich zudem immer schon vorher bewusst, bei welchem Verbrauch an Maßlösung Sie mit dem Endpunkt der Titration rechnen dürfen, um rechtzeitig – aber nicht unnötig früh – vom stetigen zum langsamen Eintropfen überzugehen.
Wichtig: Führen Sie, um ein möglichst exaktes Ergebnis zu erhalten, immer mehrere Titrationen unter den gleichen Bedingungen durch und bilden Sie den Mittelwert aus den einzelnen Messwerten. Weicht ein Ergebnis deutlich von den anderen ab, sollten Sie dieses nicht in Ihre Berechnung mit einbeziehen. Vielleicht noch wichtiger: Titrieren Sie unbedingt eine bekannte Vergleichsprobe, um den zu erwartenden Farbumschlag am Äquivalenzpunkt genau kennenzulernen.
Die erste Titration in diesem Vorkus dient zur Titerbestimmung der Natronlauge aus dem vorhergehenden Versuch. Um den Titer der Natronlauge zu bestimmen, gibt es zwei Möglichkeiten. Man kann die Natronlauge mit einer bereits eingestellten Säure titrieren oder man setzt die Natronlauge mit der Lösung eines geeigneten Urtiters um. Zum Einstellen von Natronlauge benutzt man im Allgemeinen Kaliumhydrogenphthalat als Urtitersubstanz.
Stellen Sie, um den Faktor der Natronlauge aus dem vorhergehenden zu bestimmen, eine ca. 0,05 m Kaliumhydrogenphthalat-Lösung her. Trocknen Sie dazu das Kaliumhydrogenphthalat im Trockenschrank vor (ca. 30 min bei 110 °C) und wiegen Sie die benötigte Menge (wie viel?) möglichst genau ein (notieren Sie den genauen Wert!). Überführen Sie die eingewogene Menge in einen 250-mL-Messkolben und befüllen Sie diesen zunächst etwa zur Hälfte mit destilliertem Wasser. Lösen Sie das Kaliumhydrogenphthalat durch Schütteln des Messkolbens und füllen Sie dann bis zur Eichmarke auf. Füllen Sie nun mit einer Vollpipette 25 mL der Urtiterlösung in einen Erlenmeyerkolben, verdünnen Sie mit destilliertem Wasser auf ca. 100 mL und setzen Sie einige Tropfen Phenolphthalein als Indikator zu. Füllen Sie Ihre Bürette mit der einzustellenden Natronlauge und titrieren Sie bis zum Farbumschlag nach leicht rosa.
Führen Sie die Titration mindestens dreimal durch und errechnen Sie aus dem Verbrauch der NaOH-Lösung deren Konzentration.
Pipettieren Sie mit einer Vollpipette 25 mL Ihrer Probenlösung in einen Erlenmeyerkolben, verdünnen Sie auf ca. 100 mL und durchmischen Sie das Ganze durch Schwenken des Kolbens. Geben Sie ein paar Tropfen Methylorange hinzu und titrieren Sie mit der im vorherigen Versuch eingestellten Natronlauge bis zum Farbumschlag von rot nach gelb. Notieren Sie den Verbrauch an Maßlösung und berechnen Sie die Gesamtmenge an HCl in der Probenlösung. Führen Sie die Titration mindestens dreimal durch.
Weshalb sind für die Titration von Salzsäure mit Natronlauge alle Indikatoren mit Umschlagspunkten zwischen pH 4 und pH 10 geeignet, für die Titration von Essigsäure mit Natronlauge jedoch nicht?
Die Titration einer mehrprotonigen Säure soll am Beispiel von Phosphorsäure, H3PO4, behandelt werden. Als dreiprotonige Säure besitzt die Phosphorsäure drei Äquivalenzpunkte, wobei jedoch nur die ersten beiden analytisch verwertbar sind. Eine direkte Titration auf den dritten Äquivalenzpunkt ist aufgrund der kleinen Säurestärke nicht möglich.
Pipettieren Sie mit einer Vollpipette 25 mL Ihrer Probenlösung in einen Erlenmeyerkolben. Versetzen Sie nun die unverdünnte Probe mit 3 Tropfen Methylorange und titrieren Sie die Probe mit der eingestellten Natronlauge unter ständigem Umschwenken bis zum Farbumschlag von rot nach gelb (mindestens drei Titrationen).
Pipettieren Sie mit einer Vollpipette 25 mL Ihrer Probenlösung in einen Erlenmeyerkolben. Die unverdünnte Lösung wird kurz aufgekocht und anschließend rasch unter fließenden Wasser auf Raumtemperatur abgekühlt. Es werden 3 Tropfen Phenolphthalein dazugegeben und unter Umschwenken mit der eingestellten Natronlauge bis zum ersten erkennbaren Farbumschlag nach schwach rosa titriert. (Der Farbton sollte wenigstens 30 Sekunden bestehen bleiben.)
Verwenden Sie zur Bestimmung der Phosphorsäuremenge in Ihrer Probelösung die Ergebnisse des ersten Äquivalenzpunktes. Prüfen Sie, ob der Laugeverbrauch beim zweiten Versuch doppelt so groß wie beim ersten ist.
Falls die verbrauchten Laugemengen nicht übereinstimmen – haben Sie eine Erklärung?