ca. 14 SWS (10 SWS Praktikum, 2 SWS Seminar, 2 SWS Vorlesung)
Beginn: Donnerstag, erste Vorlesungswoche,
Ende: Freitag, letzte Vorlesungswoche.
Ort: üblicherweise Praktikumssaal M in Haus D, andere Räumlichkeiten werden bei den Angaben zu den Versuchen erwähnt.
Der Klausurtermin wird zur Vorbesprechung bekanntgegeben.
Studierende des vertieften Lehramtstudiengang Chemie. Die Zugangsvoraussetzungen müssen ausnahmslos erfüllt sein.
Ziel des Praktikums ist es, Sie mit fortgeschrittenen präparativen und analytischen Methoden der Chemie vertraut zu machen. Präparativ liegen die Schwerpunkte auf Versuchen aus der Festkörperchemie sowie der Koordinationschemie. Die überwiegend zum Einsatz kommenden analytischen Methoden sind die Pulverdiffraktometrie sowie die NMR-, IR- und UV/VIS-Spektroskopie. Auch quantenmechanische Rechnungen an einfachen Systemen werden durchgeführt.
Unter der Voraussetzung, dass nur ein Praktikumssaal zur Verfügung steht und dass mehr als 18 Studenten am Praktikum teilnehmen, gilt folgendes: Das Praktikum wird in zwei Kursen durchgeführt. Die Hälfte aller Gruppen führt ihre Versuche immer Montags von 13:30–18:30 Uhr sowie Mittwochs von 13:00–18:00 Uhr durch. Die andere Hälfte hat immer Donnerstags von 14:00–18:00 Uhr sowie Freitags von 9:00–15:00 Uhr Praktikum.
Wenn maximal 18 Studenten am Praktikum teilnehmen, findet für alle das Praktikum immer Montags von 13:30–18:30 Uhr sowie Mittwochs von 13:00–18:80 Uhr statt.
Sollte ein zweiter Praktikumssaal zur Verfügung stehen, dann findet für alle das Praktikum immer Donnerstags von 13:30–17:30 Uhr sowie Freitags von 9:30–15:30 Uhr statt.
Begleitend zum Praktikum gibt es eine Vorlesung, die von Prof. Klüfers gehalten wird, und ein Seminar, das von Dr. Mayer und Prof. Karaghiosoff durchgeführt wird. Zeit und Ort der Veranstaltungen werden rechtzeitig bekanntgegeben.
Die Versuche werden in der Regel in Zweiergruppen durchgeführt. Der unterschiedliche Betreuungsaufwand durch Assistenten und Tutoren bei den einzelnen Versuchen in Verbindung mit der begrenzten Anzahl an zur Verfügung stehenden Assistenten und Tutoren, erlaubt es nicht, das Praktikum so zu organisieren, dass immer alle Gruppen zeitgleich den gleichen Versuch bearbeiten. Das hat zur Folge, dass Sie in der Regel die Versuche nicht in der Reihenfolge bearbeiten können, wie sie nachfolgend beschrieben werden. Eine weitere Konsequenz ist, dass Sie gerade zu Beginn des Praktikums noch nicht in der Lage sein werden, die Ergebnisse der analytischen Messungen auszuwerten. Umso wichtiger sind eine saubere Protokollführung sowie ein achtsames Umgehen mit den Ergebnissen der analytischen Messungen, so dass Sie zu einem späteren Zeitpunkt, wenn alle Kenntnisse und Unterlagen zu einem Versuch vohanden sind, in der Lage sind, dem Assistenten ein vollständiges Protokoll zur Korrektur und Bewertung vorzulegen.
Prüfen Sie hier, ob Ihr Browser das Skript korrekt darstellt.
Um das Skript auszudrucken, verwenden Sie am Besten die pdf-Version. Das Skript wird derzeit erstellt und kann sich während des Praktikums auch noch ändern. Bei Änderungen werden Sie vom Praktikumsleiter informiert.
Bei der Sicherheitsunterweisung zu Beginn des Praktikums erhalten Sie eine allgemeine Arbeitsanweisung, die Sie zu beachten und unterschrieben zurückzugeben haben. Im Internet finden Sie weitere hifreiche Informationen zur Arbeitssicherheit und zu Betriebsanweisungen: die GUV-R 120 Laboratorien und die Laboratoriumsordnung des Departments.
Als anschauliche Einführung liegt eine Broschüre der Gesetzlichen Unfallversicherung vor, der unter der Bestellnummer GUV-I 8553 herausgegebenen Einführung für Studierende mit dem Titel Sicheres Arbeiten in chemischen Laboratorien.
Für die analytischen Messungen, die Sie nicht selbst durchführen, müssen Sie einen Messauftrag ausfüllen, also für NMR-Spektroskopie (Haus D), Massenspektrometrie, Röntgenanalytik (Pulver), Röntgenanalytik (Einkristall), Elementaranalytik (CHNS) und Elementaranalytik (ICP/AAS). Kein Messauftrag ist nötig für die von Ihnen selbst durchgeführten IR- und UV/VIS-Messungen. Um einen Messauftrag zu erteilen, melden Sie sich unter ChemABS mit Kennung und Passwort an (wird Ihnen vom Praktikumsleiter oder den Asistenten mitgeteilt).
Dann klicken Sie in der linken Spalte unter 'Analysenaufträge' auf 'Neue Probe'. Im sich dann öffnenden Formular tragen Sie die Probenbezeichnung ein, am besten als verbindung_analysemethode_gruppennummer, geben den Lagerort sowie die zu erwartende Molekülmasse und Summenformel an und machen die nötigen Angaben zu Aggregatzustand, Eigenschaften und Empfindlichkeiten. Unter Bemerkung können Sie beispielsweise angeben, dass Sie die (restliche) Substanz nach der Messung nicht entsorgt werden soll. Zuletzt wählen Sie dann die gewünschte Analysemethode aus und drücken 'Weiter'.
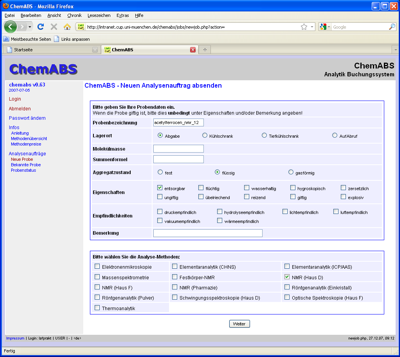
Daraufhin öffnet sich ein Formular, dessen Inhalt von der zuvor gewählten Analysemethode abhängt. In diesem Beispiel soll ein Messauftrag für die NMR-Spektroskopie in Haus D erteilt werden. Dazu tragen Sie im oberen Teil die Röhrchennummer ein (dazu später mehr), geben das Lösungsmittel an und machen Angaben zu den zu messenden Kernen. Für 1H- und 13C-Messungen können Sie die nachfolgend zu sehenden Angaben jeweils übernehmen. Im unteren Teil des Formulars (nicht abgebildet) drücken Sie dann 'Weiter'.
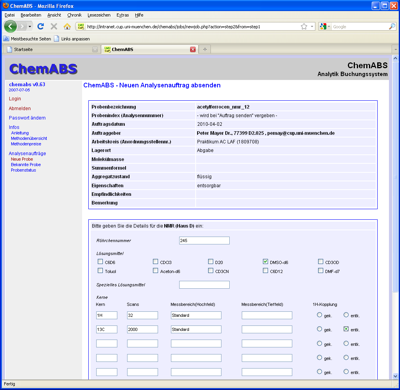
Daraufhin erscheint ein Fenster, in dem Sie die von Ihnen gemachten Angaben nochmals kontrollieren können. Möchten Sie etwas ändern, drücken Sie 'zurück', möchten Sie den Auftrag unverändert lassen, drücken Sie 'Auftrag absenden und drucken'. Tun Sie letzteres, öffnet sich ein weiteres Fenster mit den von Ihnen gemachten Angaben. Sie können dort unter dem Menüpunkt Datei die Datei auf einem geeigneten Drucker ausdrucken und danach das Fenster schließen. Im anderen Fenster drücken Sie 'Weitere Probe eingeben', wenn Sie noch einen Messauftrag erteilen möchten, oder aber Sie melden sich in der linken Spalte unter 'Abmelden' ab.
Den ausgedruckten Analysenauftrag geben Sie dann zusammmen mit der Probe am richtigen Ort ab. Für die Massenspektrometrie ist dies der Raum D0.038, für Röntgenanalytik (Einkristall) D2.025. Für Röntgenanalytik (Pulver) geben Sie Messauftrag und Probe am besten dem zuständigen Assistenten, der sich dann darum kümmert. NMR-Proben und Auftragszettel werden auf dem Probentisch im Flur im Erdgeschoss in Haus D abgegeben. Wichtig ist hierbei, dass auf dem Auftragszettel und dem NMR-Röhrchen Aufkleber mit den gleichen Nummern sind (einen dritten Aufkleber können Sie im Laborjournal einkleben. Dies ermöglicht Ihnen und dem Personal die genaue Zuordnung der Probe zum Auftrag und Versuch. Bei Unklarheiten wenden Sie sich an die Assistenten. Schreiben Sie auf den Auftragszettel auch noch LAF. Beachten Sie auch folgendes: Das NMR-Röhrchen muss wenigstens mit soviel Lösung gefüllt sein, dass die Füllhöhe etwa 5 cm beträgt. Sie benötigen also in etwa 0,6 mL (deuteriertes) Lösungsmittel! Die Lösung muss klar sein! Günstig ist auch, wenn Sie das Röhrchen und den Auftragszettel bis 15 Uhr 30 auf dem Probentisch abgeben, so dass die Probe noch zu den Nachtmessungen aufgenommen werden kann.
Ob Ihr Messauftrag bereits bearbeitet wurde oder nicht, können Sie unter ChemABS prüfen, indem Sie sich dort einloggen und
den Status Ihres Messauftrages abfragen. Wenn die Probe gemessen wurde, holen Sie sich einen Ausdruck mit dem Ergebnis ab
(gilt für Massenspektrometrie, Röntgenanalytik (Einkristall) und Elementaranalytik. Für Röntgenanalytik (Pulver) und die
NMR-Spektroskopie werden Dateien angelegt, die sie sich runterladen können. Lassen Sie sich von den zuständigen Assistenten
zeigen, wie Sie an die Dateien zur Pulverdiffraktometrie kommen. An die NMR-Dateien gelangen Sie wie folgt:
Loggen Sie sich über SSH auf der cicum4 ein (Passwort und Kennung erfahren Sie von den Praktikumsassistenten). Gehen Sie auf der cicum4 in das Verzeichnis /nmr und dann in das Verzeichnis j270 oder j400.
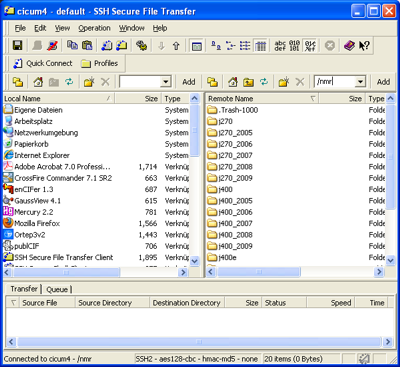
Die Namen stehen für zwei verschiedene Messgeräte. Anhand der vom Operator auf dem Auftragszettel notierten Dateibezeichnung (NMR-Probe und Auftragszettel nach der Messung wieder am Probentisch im Flur in Erggeschoss in Haus D abholen!) können Sie auf das Messgerät schließen: sind im Dateinamen zwei Buchstaben, liegen Ihre Dateien unter j270, bei drei Buchstaben liegen sie unter j400. Wählen Sie also das richtige Verzeichnis, gehen dann in das Verzeichnis jeol_files, dann in das Verzeichnis LAF und wählen dort Ihre Dateien aus, drücken die rechte Maustaste und wählen Download (zuvor das richtige lokale Verzeichnis auswählen (Assistent!)).
Wie diese Rohdaten dann weiterbearbeitet werden, wird unten beschrieben.
Sie können IR- und UV/VIS-Messungen auch in F1.058 selbständig durchführen. Der Raum ist von 8:00 – 16:00 Uhr für Studenten geöffnet. Beachten Sie bei der Durchführung der Messungen die dort ausliegenden Anleitungen.
Messungen am UV/Vis-Spektrometer in D1.023
Das Spektrometer ist in der folgenden Abbildung links zu sehen. Dazu gehört auch der links stehende Rechner mit Zubehör.

Starten Sie den PC, und schalten Sie das UV/Vis-Spektrometer ein (blauer Schalter an der rechten Seite).
Starten Sie dann das Programm Spectra Manager (Icon auf Desktop). Klicken Sie dann im Progammfenster links unter UV/Vis einmal auf Spectra Measurement und anschliessend einen Doppelklick auf die rechts stehende Methode LAF.uvsp.
Zunächst wird die Baseline gemessen. Dazu füllen Sie eine Einwegküvette mit dem Lösungsmittel, in dem sich Ihre später zu messende Substanz befindet. Platzieren Sie die Küvette im Strahlengang. Achten Sie dabei darauf, dass sich die trüben Seiten der Küvette links und rechts befinden (von vor dem Gerät stehenden Betrachter aus gesehen). Die beiden folgenden Abbildungen sollen dies verdeutlichen:


Menu: Measure - Baseline - ok.
Dann entsorgen Sie das Lösungsmittel aus der Küvette fachgerecht und füllen die gleiche Küvette mit einer Lösung, die aus dem zuvor gemessenen Lösungsmittel und der darin gelösten zu messenden Substanz besteht. Platzieren Sie diese Lösung wie oben beschrieben im Strahlengang.
Menu: Measure - Sample - Sample Name eingeben - ok.
Das Spektrum öffnet sich in einem Fenster mit dem namen Spectra Analysis. Dort wählen Sie das Menu: Processing - Peak processing - peak find. Lassen Sie dann den Wert für upper limit unverändert, lower limit klicken Sie weg, noise 0.01, wenn dann alles ok ist: Menu: Print. Speichern Sie die Datei unter einem geeigneten Namen (Gruppennummer und Versuchnummer enthalten) ab unter C:\UV/Vis-Daten+Parameter\UVVIsDaten\LAF. Die Daten können Sie auf einen hoffentlich virenfreien USB-Stick übertragen.
Speichern Sie das Spektrum dann noch beim Schliessen des Spectra Analysis Fensters als jws-datei mit geeignetem Namen ab.
Auch Festkörper können mit diesem Gerät gemessen werden. Dazu brauchen Sie einen Probenträger, dessen Einzelbestandteile in der folgenden Abbildung zu sehen sind: eine UV-Glasplatte, zwei Kunststoffbauteile und drei Metallbauteile.

Sollte der Probenträger zusammengebaut und ohne Substanz sein, können Sie ihn unverändert zur Messung der Baseline verwenden (wie oben für Lösungen beschrieben). Danach bauen Sie den Probenträger vorsichtig so weit auseinander, dass Sie ihre Substanz einbringen können. Nachfolgend wird beschrieben, wie Sie einen komplett zerlegten Probenträger zusammenbauen und mit der zu messenden Substanz füllen.
Legen Sie dann die Metallplatte auf eine ebene Fläche und platzieren Sie in der Bohrung die Glasplatte. Darauf legen Sie das flache Kunststoffbauteil mit der größeren Fläche auf die Glasplatte. Bringen Sie dann eine Spatelspitze Substanz in die Bohrung in dem Kunststoffteil.

Schrauben Sie dann das Metallgewinde auf die Platte auf.

Setzen Sie dann den Kunststoffstempel passend ein und schrauben Sie die Überwurfmutter auf, so dass die Substanz gut zusammengepresst wird.

Ihr Probenträger samt Substanz müsste etwa so aussehen, wie auf dem folgenden Bild.

Setzen Sie den Probenträger dann dort ein, wo Sie auch die Küvetten einsetzen.

Achten Sie dabei darauf, dass der Probenträger gerade und mittig eingesetzt wird. Die weisse Linie auf dem Probenträger sollte eine Gerade mit der Linie auf dem Gehäuse bilden.

Die Messung von Festkörpern erfolgt genau so wie oben für Lösungen beschrieben. Der einzige Unterschied ist, dass Sie zur Messung der Baseline den leeren Probenträger ohne Substanz messen.
Spektren aus Lösung am Cary 50 in D2.062 oder in D2.004
Tragen Sie sich in die ausliegende Liste am Spektrometer ein.
Dann starten Sie den PC am Cary 50 (falls er nicht schon gestartet ist). Kennung und Passwort soll ein Mitarbeiter der Arbeitsgruppe Klüfers eingeben.
Doppelklick auf das Icon Scan.
Dann Setup auswählen und dort unter Cary den X Mode (also den Messbereich) von 800 nm bis 200 nm einstellen, die Scan controls auf simple und medium setzen, unter Baseline Baseline correction anklicken, unter Reports bei options Parameter anklicken, dann mit ok raus.
Dann füllen Sie eine 1cm-Quarzglasküvette (Bezeichnung QS...) nur mit dem Lösungsmittel, in dem später ihre feste Verbindung gelöst werden soll, und achten dabei darauf, dass in der Küvette keine Luftblasen sind. Die Küvette nur auf den matten Seiten anfassen.
Das Cary 50 öffnen (grüner Schieber) und die Küvette einsetzen mit den matten Seiten nach vorn und hinten, den klaren Seiten nach links und rechts. Cary 50 schließen.
Baseline drücken, ok. Das Spektrum der Baseline ist nicht am Bildschirm zu sehen! Die Messung des reinen Lösungsmittels dient dazu, dessen Auswirkungen auf das UV-Spektrum zu messen und später vom Spektrum der gewünschten Verbindung wieder herauszurechnen.
Sobald die Messung der Baseline beendet ist, unter File den Menupunkt Save method as auswählen und dann in das Verzeichnis Austausch/Laf_UV_VIS/Cary50 wechseln und dort die baseline unter baseline_jahrmonattag_gn_lm.msw speichern (also z:B: baseline_100304_11_h2o.msw für eine Messung von Wasser, die am 4. März 2010 von der Gruppe 11 durchgeführt wurde).
Dann die gesamte Software schließen (immer, bevor eine neue Substanz gemessen wird).
Dann die Software neu starten mit Doppelklick auf das Icon Scan.
Unter File den Menupunkt open Method auswählen und die zuvor gespeicherte Datei baseline_jahrmonattag_gn_lm.msw laden.
Dann stellen Sie die von den zu messenden Verbindungen Lösungen in der bei den jeweiligen Versuchen angegebene Menge und Konzentration her. Achten Sie darauf, dass die Lösungen klar und nur schwach gefärbt sind. Füllen Sie dann die Quarzglasküvette mit der zu untersuchenden Lösung und achten Sie wieder darauf, dass keine Luftblasen in der Küvette sind, und dass die Küvette nur auf den matten Seiten angefasst wird. Setzen Sie die Küvette wie oben bereits beschrieben in die Halterung im Cary 50 ein und schließen Sie den Schiebedeckel.
Im Programmfenster wählen Sie dann Setup und dort tragen Sie unter der Seite Reports bei Name ihren Namen ein und geben unter comments folgendes ein: Schichtdicke d = 1 cm, Konzentration der Lösung (z.B. 0,5 mM), Name der Verbindung (z.B. Co-glycin-fac). Dann raus über ok.
Dann drücken Sie Start und wählen im sich öffnenden Fenster das Dateiformat *.bsw und geben den Filenamen ein als verbindung_gn.bsw (also z.B. co-glycin-fac_11.bsw für das fac-Isomer von Co-glycin der Gruppe 11). Drücken Sie save und geben Sie dann als sample name ebenfalls verbindung_gn ein (also z.B. co-glycin-fac) und drücken Sie ok. Sie können dann auf dem Bildschirm die Messung des UV-Spektrums ihrer Lösung verfolgen.
Sobald das Spektrum vollständig gemessen wurde, belabeln Sie die Maxima. Dazu wählen Sie das peak labels icon (2. von rechts) und wählen x and y labels. Sollten zuviele Peaks belabelt werden, ändern Sie den threshold von 0,1 auf z. B. 0,5 (einfach ausprobieren). Daraufhin drücken Sie das scale graph icon (4. von links) und ändern dort die dargestellten Bereiche der Abszisse und Ordinate. In der Regel liegen die gewünschten Maxima zwischen 300 nm und 700 nm. Die Abs-Skala wählen Sie so, dass das Maximum des intensivsten Übergangs etwa 80% der Abs-Skala erreicht.
Wenn die peaks beschriftet sind und der dargestellte Bereich ok ist, speichern Sie das ganze. Save data as unter zuvor gewähltem Dateinamen verbindung_gn.bsw.
Dann drücken Sie im Reportfenster unterhalb des Fensters, in dem das Spektrum abgebildet ist, die rechte Maustaste und wählen edit. Dann löschen Sie alle Leerzeilen im Report und speichern danach wie gewohnt unter save data as als verbindung_gn.bsw.
Dann drucken Sie das Spektrum in eine pdf-datei unter dem Namen verbindung_gn.pdf. Diese können Sie dann auf einem USB-Stick mit nach Hause nehmen und auswerten.
Dann schließen Sie das Programm.
Die Küvetten bitte mit dem Lösungsmittel reinigen, das zuletzt verwendet wurde. Die Lösungsmittel dann bitte sachgerecht entsorgen!
Haben Sie eine weitere Verbindung zu messen, starten Sie wie oben beschrieben das Programm erneut und beginnen mit open Method...
Tragen Sie in der ausliegenden Liste noch die Uhrzeit zum Ende der Messungen ein.
Festkörperspektren am Cary 500 in D2.044
Tragen Sie sich in die ausliegende Liste am Spektrometer ein.
Dann starten Sie den PC am Cary 500 (falls er nicht schon gestartet ist). Kennung und Passwort soll ein Mitarbeiter der Arbeitsgruppe Klüfers eingeben.
Doppelklick auf das Icon CaryWinUV.
Dann Doppelklick auf das Icon Scan Shortcut. Unten links im sich öffnenden Fenster steht, in welchem Zustand sich das Cary 500 befindet. Steht dort Cary offline, dann schalten Sie das Cary 500 ein (Kippschalter links unten am Cary 500). Nach wenigen Minuten ist der Zustand des Cary 500 Idle. Dann lassen Sie das Gerät etwa 30 Minuten warm laufen.
In der Zwischenzeit kleben Sie die zwei Quarzglasplatten (aus dem LAF/ACII-Praktikum mitbringen) auf gegenüberliegenden Seiten mit etwas Tesafilm zusammen. Kennzeichnen Sie die oben liegende Platte mit einem V.

Öffnen Sie das Cary 500.

Setzen Sie die Quarzglasplatte so ein, dass die mit V gekennzeichnete Platte nach vorn zeigt.

Wenn Sie die Quarzglasplatten in die richtige Position über dem Gummistopfen gebracht haben, drücken Sie die schwarze Halterung dagegen, so dass die Quarzglasplatten zwischen Gehäuse und schwarzer Halterung eingeklemmt werden.

Schließen Sie die Probenkammer des Cary 500.

Zur Messung der Baseline geht man vor wie am Cary 50. Setup auswählen und dort unter Cary den X Mode (also den Messbereich) von 800 nm bis 200 nm einstellen, Y Mode auf %R einstellen, unter Baseline Baseline correction anklicken, unter Reports bei options Parameter anklicken, dann mit ok raus.
Baseline drücken, ok. Die Messung der leeren Glasplatten dient dazu, deren Auswirkungen auf das UV-Spektrum zu messen und später vom Spektrum der gewünschten Verbindung wieder herauszurechnen.
Sobald die Messung der Baseline beendet ist, unter File den Menupunkt Save method as auswählen und dann in das Verzeichnis Austausch/Laf_UV_VIS/Cary500 wechseln und dort die baseline unter baseline_jahrmonattag_gn.msw speichern (also z:B: baseline_100304_11.msw für eine Messung, die am 4. März 2010 von der Gruppe 11 durchgeführt wurde).
Dann dieses Fenster schließen (immer, bevor eine neue Substanz gemessen wird).
Dann die Software neu starten mit Doppelklick auf das Icon Scan Shortcut.
Unter File den Menupunkt open Method auswählen und die zuvor gespeicherte Datei baseline_jahrmonattag_gn.msw laden.
Dann öffnen Sie die Probenkammer des Cary 500 und nehmen die Quarzglasplatten raus. Halten Sie die Platten gut fest! Lösen Sie auf einer Seite den Tesafilm und klappen Sie die Platten auseinander. Bringen Sie eine kleine Spatelspitze ihrer zu messenden Substanz in die Mitte einer Glasplatte.

Verteilen Sie die Substanz mit dem Spatel ein bisschen.

Klappen Sie leere Glasplatte wieder auf die mit Substanz beschichtete und verteilen Sie die Substanz gleichmäßig zwischen den beiden Platten, indem Sie die beiden Platten eine Weile leicht gegeneinander verschieben. Wenn Sie eine gleichmäßige Verteilung erreicht haben (eine lückenlose werden Sie kaum schaffen), kleben Sie die beiden Platten wieder zusammen.

Setzen Sie dann die Quarzglasplatten, wieder wie oben beschrieben in der Probenkammer ein. Achten Sie dabei darauf, dass die mit V gekennzeichnete Platte wieder nach vorn zeigt. Schließen Sie die Probenkammer.
Im Programmfenster wählen Sie dann Setup und dort tragen Sie unter der Seite Reports bei Name ihren Namen ein und geben unter comments den Namen der Verbindung ein. Dann raus über ok.
Dann drücken Sie Start und wählen im sich öffnenden Fenster das Dateiformat *.bsw und geben den Filenamen ein als substanzname_gn.bsw (also z.B. kaliumsulfat_11.bsw für die Gruppe 11). Drücken Sie save und geben Sie dann als sample name ebenfalls substanzname_gn.bsw (also z.B. kaliumsulfat) und drücken Sie ok. Sie können dann auf dem Bildschirm die Messung des Reflexionsspektrums ihrer Substanz verfolgen.
Wenn die Messung des Spektrums abgeschlossen ist, muss es in ein Absorptionsspektrum umgerechnet werden. Klicken Sie dazu auf das Icon, auf dem ein Taschenrechner abgebildet ist. Dort wählen Sie selected trace, convert to F(R), und selected graph, drücken dann den apply button und die = Taste. Dann erscheint der Graph des Absorptionsspektrums im gleichen Display wie das Reflexionsspektrum. Wählen Sie anstelle von selected graph new graph, erscheint das Absorptionsspektrum allein.
Am Graphen des Absorptionsspektrums sollte dann wieder der Messbereich angepasst werden und die Maxima beschriftet werden. Klicken Sie dazu auf den Graphen des Absorptionsspektrums, so dass er in roter Farbe abgebildet ist. Dann wählen Sie das peak labels icon (2. von rechts) und wählen x and y labels. Sollten zuviele Peaks belabelt werden, ändern Sie den threshold von 0,1 auf z. B. 0,5 (einfach ausprobieren). Daraufhin drücken Sie das scale graph icon (4. von links) und ändern dort die dargestellten Bereiche der Abszisse und Ordinate. In der Regel liegen die gewünschten Maxima zwischen 300 nm und 700 nm. Die Abs-Skala wählen Sie so, dass das Maximum des intensivsten Übergangs etwa 80% der Abs-Skala erreicht.
Wenn die peaks beschriftet sind und der dargestellte Bereich ok ist, speichern Sie das ganze. Save data as unter zuvor gewähltem Dateinamen substanzname_gn.bsw.
Dann drucken Sie das Spektrum in eine pdf-datei unter dem Namen substanzname_gn.pdf. Diese können Sie dann auf einem USB-Stick mit nach Hause nehmen und auswerten.
Dann schließen Sie das Fenster.
Haben Sie eine weitere Verbindung zu messen, starten Sie wie oben beschrieben das Programm erneut und beginnen mit open Method...
Nehmen Sie die Quarzglasplatten aus der Probenkammer raus und reinigen Sie sie mit einem feuchten Papier. Das beschmutzte Papier nehmen Sie anschließend mit und entsorgen es in der Feststofftonne.
Tragen Sie in der ausliegenden Liste noch die Uhrzeit zum Ende der Messungen ein.
Messungen am IR-Spektrometer in D1.023
Das IR-Spektrometer ist das rechte der beiden in der folgenden Abbildung zu sehenden Geräte. Dazu gehört der über dem Gerät stehende Computer mit Monitor. Die Maus dazu befindet sich zwischen den beiden Spektrometern, die Tastatur auf den beiden Spektrometern.
Fahren Sie den PC hoch und schalten Sie dann das IR-Spektrometer ein (Power on). Nachdem es dreimal gepiepst hat, ist das Spektrometer betriebsbereit.
Doppelklick auf Icon Spectra Manager. Dann in der linken Spalte auf FT-IR-Photometer klicken, dann auf Spectra Measurement klicken, und dann Doppelklick auf LAF.par.
1. Background measurement, Menu Measure - Background auswählen, Sample Name eingeben (sollte Gruppennummer und Versuchsnummer beinhalten), OK. Die Background Messung erfolgt bei weggedrehtem Stempel und offenem Gehäuse (siehe folgende Abb.).

2. Dann wird die Probe aufgebracht (eine kleine Spatelspitze reicht).

Drehen Sie am schwarzen Drehknopf den Stempel so hoch, dass Sie ihn ohne Probleme über die Probe bringen können.

Dann drehen Sie am grauen Drehknopf den Stempel auf die Probe, nicht zu fest.

3. Menu: Measure - Sample - Sample Name eingeben - OK.
4. Nach der Messung öffnet sich das Spektrum in einem eigenen Fenster mit dem Namen Spectra Analysis.
5. Menu: Processing - Peak processing - peak find. Es öffnet sich ein neues Fenster. Dort Noise level auf 0.5 setzen, dann upper und lower limit so wählen, dass nach Drücken von Apply die gewünschten Peaks erkannt werden. Unerwünscht erkannte Peaks können durch anklicken in der Liste links gelöscht werden. Dazu rechte Maustaste drücken und Delete wählen.
6. Wenn alles ok: Menu Print - Print. Speichern unter: c:\FTIR-Daten+Parameter\FTIR-daten\LAF. Dateinamen eingeben (sollte Gruppennummer und Versuchsnummer enthalten), PDF wird erzeugt. Die Datei kann dann auf einem mitgebrachten USB-Stick gespeichert werden. Bitte saubere (virenfreie) USB-Sticks verwenden!
7. Dann im Spectra Analysis Fenster: Menu: File - Save.
8. Spectra Measurement Fenster schliessen.
9. Stempel hoch- und wegdrehen. Mit feuchtem Papier den Feststoff sauber abputzen. Bitte auch den Stempel von unten mit feuchtem Papier putzen.
10. Wenn das Gerät nicht mehr gebraucht wird: Stempel so weit nach unten drehen, dass das Gehäuse geschlossen werden kann.
11. Software schliessen und Power off drücken.
Messungen am IR-Spektrometer in D2.048
Tragen Sie sich in die ausliegende Liste am Spektrometer ein.
Schalten Sie das IR-Gerät ein (grüner Schalter hinten rechts neben dem Computer).
PC hochfahren und Monitor einschalten. Ein Mitarbeiter des AK Klüfers wird sich einloggen. Warten Sie, bis Sophos Ruhe gibt (Symbol links neben der Uhrzeit rechts unten am Bildschirm).
Doppelklick auf Icon Spectra Manager. Dann im sich öffnenden Fenster Doppelklick auf Spectra Measurement. Wenn dann die Meldung kommt, dass keine Verbindung zum Spektrometer aufgebaut werden konnte, alles runterfahren und ausschalten und in der oben beschriebenen Reihenfolge neu starten.
Wenn Verbindung aufgebaut werden kann, erscheint das Background Spektrum. Um dieses neu zu messen, wird der Messkopf in die Mitte gedreht und mit dem scharzen Drehknopf nach unten auf den Probenhalter bewegt, bis ein stärkerer Widerstand auftritt (nicht mit Gewalt weiterdrehen).

Dann in der Menuleiste auf Measure, dann Background. Warten, bis das Background-Spektrum vollständig gemessen wurde. Dann überprüft man, ob der Background ok ist, indem man einfach eine Messung ohne Substanz macht. In Menuleiste auf Measure, dann Sample. Es sollte dann ein Spektrum erscheinen, in welchem die Transmission zwischen 99% und 101% liegt. Dann ist alles ok. Ansonsten Background nochmals messen. Eine etwas höhere Intensität bei 2350 cm-1 ist kein Problem.
Dann dreht man mit dem schwarzen Drehknopf den Messkopf nach oben und dreht ihn nach rechts zur Seite.

Dann bringt man mit einem Spatel wenig Substanz in die Mitte der Probenhalterung.

Dann dreht man den Messkopf wieder in die Mitte und dreht ihn mit dem schwarzen Drehknopf nach unten über die Substanz, bis ein stärkerer Widerstand auftritt (wieder nicht mit Gewalt weiterdrehen).
Dann misst man das IR-Spektrum der Substanz und wählt in der Menuleiste Measure, Parameter + Sample. Im sich öffnenden Parameter-Fenster füllt man unter Information sample name (als Probenname wählt man substanz_gn, also z.B. NaCl_12 für Natriumchlorid der Gruppe 12) und Operator (Namen der Messenden) aus, kopiert den sample name und geht mit ok raus.
Wenn das Spektrum am Bildschirm erscheint, öffnet sich auch das Spectra analysis Fenster. Unter File, save as speichert man das Spektrum unter Z:\Austausch\LAF_IR unter dem Namen substanz_gn (zuvor kopierten sample name einfach einfügen).
Unter View, scale kann man den sichtbaren Bereich ändern (%T und cm-1).
Unter Processing, peak process, peak find kann man im sich dann öffnenden Fenster das noise level ändern und die Intensitäten und Wellenzahlen zu den peaks angeben lassen. Execute drücken und das Fenster auf maximale Größe ändern und prüfen, ob alle peaks angegeben sind. Wenn nicht: retry, noise level ändern, execute. Oder, wenn nur wenige peaks fehlen: fehlenden peak im Maximum anklicken mit rechter Maustaste und add drücken. Auf gleiche Weise können mit delete auch peaks aus der Liste gelöscht werden.
Wenn die gewünschten peak-Informationen alle da sind, print (Mitarbeiter von AK Klüfers fragen). Wenn das nicht möglich ist, in pdf-Datei drucken und auf Stick mitnehmen (umständlich, da USB-Anschlüsse am Rechner nur schwer zugänglich).
Nach der Messung alle Fenster schließen, Messkopf hochdrehen und zur Seite schieben. Mit feuchtem Papier Unterseite des Messkopfes und Probenauflagefläche reinigen. Danach mit trockenem Papier abreiben.
Nach letzter Messung Uhrzeit in Liste eintragen.
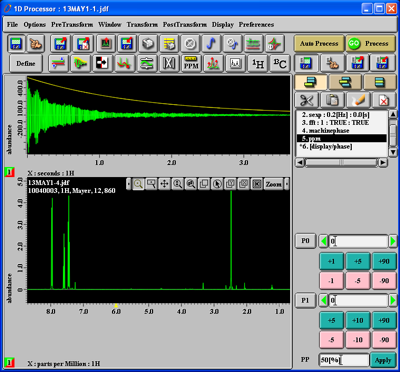
Nachdem Sie Ihre *.jdf-Rohdatenfiles von der cicum4 in Ihr lokales Verzeichnis kopiert haben, können Sie *.jdf-Datei durch Doppelklick in der Delta-Software öffnen. Sie bekommen dann etwa folgendes zu sehen:
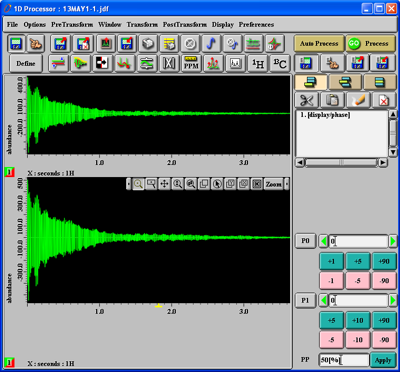
Unter den beiden Abbildungen im Fenster sehen Sie, welcher Kern gemessen wurde: hier 1H ('X:seconds:1H'). Um dieses zeitabhängige Signal zu transformieren, drücken Sie den '1H'-Button und darauf den 'Go Process'-Button. Wenn die Messung eines 13C-Kerns geladen wurde, drücken Sie entsprechend den '13C'-Button. Sie bekommen dann in etwa folgendes zu sehen (oben das zeitabhängige Signal, unten das zeitunabhängige Spektrum):
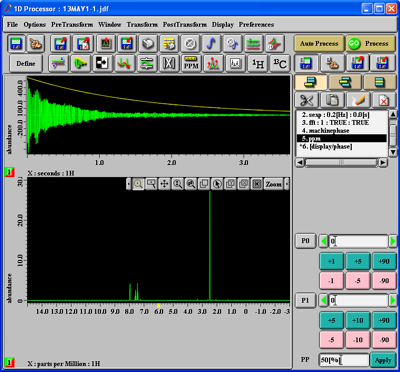
Mit der Tastenkombination 'Alt-f' können Sie den Dateinamen einblenden, mit 'Alt-g' Gitterlinien ausblenden und mit 'Alt-shift-c' die Nummer einblenden (nochmaliges Drücken der Tasten würde die Aktion rückgängig machen). Sie erhalten folgendes:
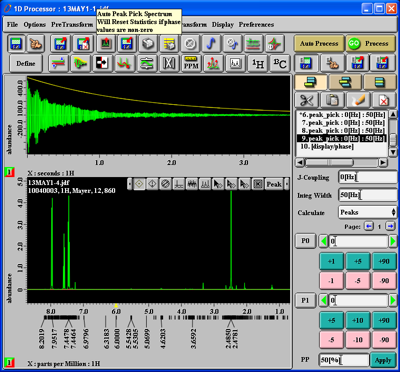
Dann drücken Sie den 'Zoom'-Button ganz rechts in der Menuleiste am oberen Rand der unteren Abbildung und wählen in der sich öffnenden Auswahlliste 'Zoom'. Ziehen Sie nun bei gedrückter linker Maustaste im Spektrum ein Rechteck um den zu vergrößernden Bildausschnitt, worauf Sie den Spektrumsausschnitt im Rechteck vergrößert erhalten:
Um die Peaks auszuwählen und zu belabeln, gehen Sie wie folgt vor: Drücken Sie wieder den 'Zoom'-Button und wählen Sie im sich öffnenden Menu 'Peak'. Dann drücken Sie den 'Auto Peak Pick'-Button unter PostTransform (X in einem Achteck). Sie erhalten:
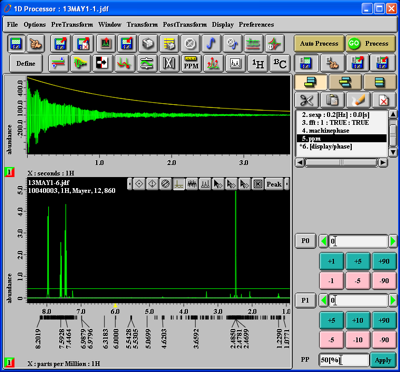
Werden zu viele Peaks belabelt, können Sie eine Feineinstellung vornehmen. Drücken Sie den 'Peak'-Button und wählen Sie in der sich öffnenden Auswahlliste 'Peak'. Dann drücken Sie in der Reihe von Buttons am oberen Rand des Spektrums den 4. Button von links. Sie können dann bei gedrückter linker Maustaste im Spektrum die grüne Linie so ziehen, dass schwache Peaks unterhalb der grünen Linie liegen:
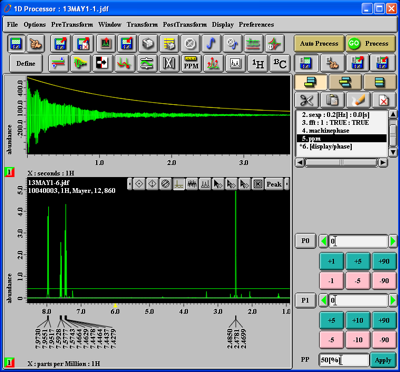
Nach erneumtem Drücken des 'Auto Peak Pick'-Buttons unter PostTransform werden die entsprechenden Peaks nicht mehr belabelt:
Um das Spektrum zu drucken, drücken Sie den Drucker-Button unterhalb von Transform:
Dann drucken Sie das Spektrum in ein File (direkt ausdrucken geht nicht!)! Stellen Sie dies im sich öffnenden Fenster ein. Wählen Sie einen geeigneten Namen für die name.eps-Datei und in dem Sie den File-Button unten links drücken ein geeignetes Verzeichnis. Wichtig: oben links muss File stehen, im Übrigen Landscape-Format, A4.
Wenn Sie im Explorer mit der rechten Maustaste auf die erzeugte name.eps-Datei drücken und in der sich öffnenden Auswahlliste 'in PDF konvertieren' wählen, können Sie die eps-datei in eine name.pdf-datei konvertieren.
1) Convert
a) Doppelklick auf Icon Convert, dann auf "open" drücken
b) in den Ordner "D:/Daten/Kurs2" wechseln und gewünschtes njc-File öffnen
c) "Save" drücken und als Dateiendung ".val" anhängen
d) Programm schließen
2) Seifert_Convert
a) Doppelklick auf Icon Seifert_Convert, dann auf "Öffnen" drücken
b) ".val"-Datei auswählen, automatisch erscheint "Datei speichern unter", "speichern" drücken (speichert als .txt-Datei)
c) Programm schließen
3) MainMenu-212
a) Doppelklick auf MainMenu-212, dann im Untermenu "Raw Data" auf "raw Data Handling" drücken
b) im Untermenu "File" auf "Import", Dateiendung "ASCII X/Y" auswählen und die erstellte ".txt"-Datei öffnen, automatisch
erscheint "Save imported file as", dort "speichern" drücken
c) falls größere Bereiche bei kleinen oder großen Beugungswinkeln keine Reflexe enthalten, können diese hier abgeschnitten werden:
im RawDat-Fenster im Untermenu "Ranges" auf "Truncate Range" gehen, da den zu behaltenden Bereich auswählen und mit "File" → "save as" die ".raw"-Datei
überschreiben
d) Rawdat schließen, im Hauptprogramm "STOE WinXPOW" im Untermenu "Raw Data" → "Graphics" auswählen
e) im "Graphics"-Fenster im Untermenu "File" auf "open..." klicken und dann die ".raw"-Datei auswählen
f) im Untermenu "Options" den Menupunkt "Background" wählen, dann auf "Subtract BG", dann im erscheinenden "Datei speichern unter"-Fenster
auf "speichern" und das Fenster über Exit schließen
g) im "STOE WinXPOW"-Fenster im Untermenu "Phase Analysis" den Menupunkt "Search/Match" auswählen
h) im "search"-Fenster über "File" und "open" die ".rmb"-Datei öffnen (dazu muss als Dateityp ".raw" ausgewählt sein)
i) im "Search"-Fenster im "Select" Untermenu auf "Elements" gehen und über "Toggle" alle Elemente entfernen, dann über die gelbe
Fläche alle Elemente markieren, die enthalten sein können, rot markierte Elemente müssen enthalten sein; in den erscheinenden
Fenstern auf "Range" und dann "Replace" drücken
j) im "Search"-Fenster kann nun über den "N"-Button die Liste aller möglichen Elemente angezeigt werden
k) die Verbindungen können nun der Reihe nach durchgetestet werden, indem der ">"-Button im Diffraktogrammfenster gedrückt wird
oder indem in der Liste drirekt ausgewählt wird
l) passende Verbindungen können über den "Select"-Button in dem Fenster, in dem das Diffraktogramm angezeigt wird, ausgewählt
werden, worauf diese Reflexlagen immer angezeigt werden, auch wenn nach anderen Elementen gesucht wird
m) im STOE WinXPOW-Fenster im RawData Untermenu Graphics auswählen und dort unter File die gewünschte .rwa-Datei öffnen
n) im sich öffnenden Graphic-Fenster den "R"-Button drücken, im view Untermenu auf Filenames
o) dann im Window Untermenu "Add ICDD Data" wählen und die passenden PDF-Nummern eingeben, die aus dem Search-Fenster
abgelesen werden können
p) dann im Graphic-Fenster File → Export Graphic wählen, das Dateiformat auswählen, z.B. .TIFF oder .BMP und einen
Namen eingeben (ohne Punkt und Endung), dann speichern
q) die gespeicherte(n) Datei(en) kann/können nun auf einem USB-Stick gespeichert und in die Protokolle eingebunden
werden.
Die Elementarzellen der im Praktikum synthetisierten Kristalle sind natürlich längst bekannt und in der Literatur
beschrieben, jedoch gehen wir davon aus, dass sie unbekannt seien, und bestimmen die Elementarzellen durch die
im folgenden beschriebene Vorgehensweise.
Ein Röntgendiffraktogramm ist dann erfolgreich indiziert, wenn für jeden Röntgenreflex mit dem zugehörigen Beugungswinkel θ
die Millerschen Indizes hkl bestimmt werden können, so dass konstante Werte für die Gitterkonstanten gefunden werden.
Für kubische Systeme gestaltet sich das Indizieren insofern einfach, dass lediglich die Gitterkonstante a bestimmt werden
muss.
Grundlage für die Indizierung kubischer Kristalle ist zum einen der Netzebenenabstand dhkl zwischen
gleichwertigen Ebenen hkl
dhkl = a × (h2 + k2 + l2)–1/2
(1)
sowie die Braggsche Gleichung
dhkl = λ × (2 sinθ)–1 (2)
mit der Wellenlänge λ der bei der Messung verwendeten Röntgenstrahlung. Einsetzen von Gleichung (1) in Gleichung (2)
gefolgt von Quadrieren ergibt
sin2θ = λ2 × (4a)–2 ×
(h2 + k2 + l2) (3).
Gesucht ist die Gitterkonstante a mit dem zugehörigen Millerschen Indextripel hkl von 100. Da die Millerschen
Indizes immer ganzzahlig sind, ist auch der Ausdruck (h2 + k2 + l2) in
Gleichung (3) immer ganzzahlig. Daraus folgt, dass alle sin2θ ganzahlige Vielfache von
λ2 × (4a)–2 sind. Hierin liegt der Schlüssel zur Bestimmung des zum Reflex 100
gehörenden Beugungswinkels θ. Legen Sie eine Tabelle wie folgt an (starten Sie mit den kleinsten Beugungswinkeln)
und berechnen Sie zu jedem θ den zugehörigen Wert für sin2θ. Die beiden anderen Spalten werden
erst später ausgefüllt.
| θ | sin2θ | ∑ | hkl |
|---|---|---|---|
| ... | ... | ... | ... |
| ... | ... | ... | ... |
| ... | ... | ... | ... |
| ... | ... | ... | ... |
Nehmen Sie nun die drei sin2θ-Werte (a), (b) und (c), die zu den drei kleinsten Beugungswinkeln θ gehören, und tragen Sie sie in folgende Tabelle ein. Dividieren Sie dann die drei sin2θ-Werte in jeder Zeile durch die in der ersten Spalte stehende ganze Zahl.
| Ganze Zahl | sin2θ (a) | sin2θ (b) | sin2θ (c) |
|---|---|---|---|
| 1 | ... | ... | ... |
| 2 | ... | ... | ... |
| 3 | ... | ... | ... |
| 4 | ... | ... | ... |
| 5 | ... | ... | ... |
Sobald Sie in allen drei Spalten eine übereinstimmende Größe gefunden haben (diese übereinstimmende Größe wird in den drei Spalten für unterschiedliche Ganze Zahlen auftreten!!), haben Sie einen möglichen Wert für sin2θ100 für den Reflex 100 gefunden. Wenn dies so ist, muss sich für alle Reflexe in der ersten Tabelle bei der Division von sin2θ durch sin2θ100 eine ganze Zahl ∑ ergeben. Dann können Sie den ganzen Zahlen Reflexe hkl zuordnen (z.B. gehört zu ∑ = 3 der Reflex 111, zu ∑ = 9 gehören 221 oder 300), auch indizieren genannt.
Die Rechnungen mit Gaussian werden im CIP-Raum in Haus D durchgeführt. Damit Sie dort arbeiten können, benötigen
Sie eine Studentenkennung cxxxx (xxxx = Nummer). Falls Sie keine haben, müssen Sie zu Beginn des Praktikums eine
Einführungsveranstaltung von Herr Dr. Wagner besuchen. Ob solch eine Veranstaltung nötig ist oder nicht, wird noch
vor Beginn des Praktikums geklärt werden.
Bei quantenmechanischen Rechnungen werden die Energie, die Masse und (Bindungs)Längen gerne in atomaren Einheiten
angegeben. Die folgende Tabelle gibt die Umrechnungsfaktoren in gebräuchlichere Einheiten an. Auch
hier können Sie Einheiten konvertieren.
| Eigenschaft | Atomare Einheit | Umrechnung |
|---|---|---|
| Länge | Bohr | 1 Bohr = 0,529177249 Å |
| Masse | atomare Masseneinheit (amu) | 1 amu = 1,6605402 × 10–27 kg |
| Energie | Hartree | 1 Hartree = 27,2116 eV |
Seit ihren Anfängen hat sich die theoretische Chemie und die Modelle für den Aufbau von Atomen und Molekülen von Demokrit über Dalton, Thompson, Rutherford und Bohr hin zum Molekülorbitalmodell soweit entwickelt, dass auf deren Grundlage Voraussagen über physikalisch-chemische Eigenschaften und Strukturen möglich sind. Vor allem die MO-Theorie stellt einen bedeutenden Grundpfeiler der modernen Bindungsvorstellung in der Molekülchemie dar.
In der unteren Abbildung sehen Sie das aus der Grundvorlesung bekannte MO-Diagramm von O2.

Wie entsteht nun ein solches MO-Diagramm? Zunächst einmal kann die MO-Theorie schlicht qualitativ angewendet werden, um die prinzipielle Abfolge der Molekülorbitale darstellen zu können, wie das Beispiel eines qualitativen MO-Schemas für ein zweiatomiges Molekül aus dem p-Block der zweiten Periode in der folgenden Abbildung.

Solche qualitativen MO-Schemata sind jedoch schon für geringfügig komplexere Moleküle (unterschiedliche Elektronegativität der Bindungspartner, unterschiedliche Valenzschalennummer, etc.) nicht mehr intuitiv ableitbar. Abhilfe schaffen hier computerchemische Rechenprogramme. Auch die Energien der Orbitale des obigen MO-Diagramms für das O2-Molekül sind mit Hilfe eines solchen Rechenprogramms ermittelt worden.
Aber nicht nur die energetische Abfolge, einhergehend mit der Ionisierungsenergie und Elektronenaffinität oder die Gestalt von Molekülorbitalen kann berechnet werden, sondern Parameter verschiedenster Art wie Bindungsabstände, Bindungswinkel, NMR-Signale, IR-/Raman- und UV-Vis-Spektren, deren Werte mit teilweise erstaunlicher Genauigkeit experimentellen Ergebnissen nahe kommen.
Die Computerchemie hat auch im Forschungsalltag ihren festen Platz als ubiquitär vorhandene Methode zur Vorhersage von Strukturen, Energien etc. gefunden und darf deshalb genauso wenig wie die Methoden von UV/VIS- /IR-, der NMR-Spektroskopie und Röntgenbeugung in der fachwissenschaftlichen Ausbildung der Lehrkräfte vermisst werden, zumal durch die technische Entwicklung seit den 50iger Jahren es heut zu Tage auch mit Heim- und Schulcomputern möglich geworden ist, quantenmechanische Berechnungen an kleinen für die Schulchemie relevanten Molekülen in geeignetem Zeitrahmen durchzuführen.
Dafür benötigt man zum einen ein Programm zum Erstellen des jeweiligen Moleküls und der Darstellung der Rechenergebnisse sowie ein Programm, welches die Rechenarbeit übernimmt. Im universiäteren Rahmen werden verschiedenste kostenpflichtige Softwareprodukte angeboten. Des Weiteren bieten sich für den privaten und schulischen Zweck auch kostenlose Programme an.
Im Folgenden sollen Sie die Grundlagen der quantenmechanischen Berechnung von Molekülparametern selbst erarbeiten. Zudem wird im Folgenden eine kostenlose Programmkombination zur Ausführung und Auswertung solcher Rechnungen vorgestellt, mit deren Hilfe Sie die Aufgaben von Versuch 1 bearbeiten sollen.
Machen Sie sich die folgenden Grundbegriffe anhand von weiterführender Literatur klar.
Rechenmethode (HF, RHF, UHF, DFT,...), Korrelationsmethode (MP2...), Basissatz (6-331G-2dp,...)
Das Wissen um diese Begriffe und ihren Zusammenhang sollte Ihnen einen Eindruck vermitteln können, was Sie bei Versuch 1 überhaupt tun und als grundlegendes Wissen ausreichen. Sie müssen keine mathematischen Ableitungen oder dergleichen beherrschen, sollten aber prinzipiell verstanden haben, wie Moleküle berechnet werden.
Literatur
- Riggenmann, T., Einführung in die computerchemische Betrachtung basischemischer Sachverhalte mittels frei erhältlicher Software sowie Darstellung von {FeNO}7-Komplexen mit Citrato- und Tartratoliganden, Zulassungsarbeit im Fachbereich Anorganische Chemie, 2011.
Einführung in die Grundbegriffe der Computerchemie, sowie beispielhafte Anwendung der Programmkombination Gabedit/Gamess.- Cramer, C. J., Essentials of computational chemistry: theories and models. 2nd edition, 2004: Wiley: New York.
Leicht verständliches Standardwerk zur Computerchemie. Auch ohne tieferes mathematisches Verständnis können die Aussagen der Formeln aus dem erklärenden Begleittext zusammengefasst werden.- Jensen, F., Introduction to computational chemistry. 2nd edition, 2007: John Wiley & Sons: New York.
Ebenfalls ein Standardwerk zur Computerchemie. Allerdings fehlt der zum Großteil der leich verständliche erklärende Charakter des obigen Werks.- INTERNETRECHERCHE
Im Folgenden wird nicht das gesamte Repertoire der Programmfunktionalitäten aufgezeigt werden, sondern lediglich die Anleitung vom Bauen eines Moleküls über die Rechnung hin zur Auswertung der an diesem Molekül berechneten Parameter. Scheuen Sie sich nicht, selbst mit dem Programm zu spielen und dessen Funktionsvielfalt zu entdecken, es kann einiges!!!
Zum kostenlosen Download der aktuellen Version von Gabedit gelangen Sie hier.
Mit Hilfe dieser Software können Sie Moleküle selbst zusammenbauen, sei es durch "aneinanderklicken" einzelner Atome oder bei größeren Molekülen über die Fragment-Funktion (Benzen!).
Unten sehen Sie das Startfenster in seinem Erscheinungsbild.

Durch Klicken des fünften Icons von rechts in der Navigatinsleiste Draw geometry öffnet sich ein neues Fenster, in dem das jeweilige Molekül zusammengebaut werden kann und wie folgt aussieht.

Die oberen fünf Werkzeuge in der links am Fenster befindlichen Werkzeugleiste dienen zur Veränderung des Blickwinkels auf das Molekül. Darunter sind Werkzeuge für die Veränderung und Transformation des Moleküls untergebracht, gefolgt von weiteren Möglichkeiten zur Veränderung der Darstellungsweise des Moleküls (z.B. Wechsel von Ball&Stick-Darstellung zum Drahtgittermodell). Ganz unten befindet sich die Messfunktion zur Ermittlung und Einstellung von Bindungsabständen und -winkeln.
Durch Anwählen des Stiftsymbols und Klicken auf das darunter befindliche Periodensystemsymbol gelangt man zum Fenster für die Auswahl des gewünschten Atoms.

Platziert man ein Atom auf der Draw geometry-Ebene, klickt auf das nun vorhandene Atom, hält die Maustaste gedrückt und zieht den Mauszeiger ein wenig zur Seite, entsteht ein zweiatomiges Molekül, im Beispiel O2. Denken sie an die Werkzeuge zur Änderung des Blickwinkels.

Durch Rechtsklick in der Zeichenebene gelangt man sowohl zu den aus der linken Werkzeugleiste bekannten, wie auch noch weiteren Funktionen. Beispielsweise lässt sich hier unter Operations->Insert/Change atoms or bond und anschließendem Klicken auf die Bindungsachse eine Doppelbindung erzeugen, die in der Ball&Stick-Darstellung deutlicher zu sehen ist. Des Weiteren lässt sich mit Hilfe der Messfunktion der Bindungsabstand (bei drei- oder mehratomigen Molekülen auch die Bindungswinkel) nach Wunsch einstellen. Dazu müssen die jeweiligen Atome nach Anwahl der Messfunktion der Reihe nach angeklickt werden.

Im Folgenden wird ausgehend vom gerade gebauten O2-Molekül erläutert, wie mit Hilfe von Gabedit Eingabedateien für Rechenprogramme, hier im Speziellen für Gamess, erstellt werden können.
Zunächst wird im Startfenster von Gabedit über File->New->Gamess input das Fenster für die Einstellung der Rechenparameter geöffnet und diese definiert.

Nach Klicken auf den OK-Knopf erscheint die von Gabedit erstellte Eingabedatei im Startfenster.

Unter dem Menüpunkt File kann die Eingabedatei gespeichert werden. Der Name erscheint dann auch im Startfenster.
Die Installationsdatei für das Rechenprogramm Gamess erhalten Sie über diese Homepage. Hierfür bedarf es unter Punkt GAMESS->Downloads->obtaining GAMESS einer kostenlosen Registrierung. Der Link zur Exe-Datei wird Ihnen dann innerhalb der nächsten beiden Werktage zugeschickt. Lassen Sie bei Erhalt der E-Mail nicht zu viel Zeit verstreichen, denn der mitgeschickte Login-Code für die Datei ist jeweils nur eine Woche gültig.
Für die Benutzung von Gamess muss in Gabedit unter dem Menüpunkt Settings->Preferences im Bereich Others der entsprechende Installationspfad von Gamess angegeben werden. Ist dies nicht der Fall, findet Gabedit das Rechenprogramm nicht und die Rechnung kann nicht ausgeführt werden.

Anschließend kann über den Menüpunkt Run->Run Computation Chemistry program das Fenster für den Start der Rechnung aufgerufen werden.

Es ist darauf zu achten, dass als Programm Gamess und als Host Local angewählt ist. Der Speicherort sowie der Name der Datei kann hier festgelegt werden. In der Zeile Command to execute sollte der Name der Programmdatei von Gamess stehen (z.B. gamess.11-32.exe, nachzulesen im Installationspfad von Gamess). Mit Klicken auf den OK-Knopf wird die Rechnung gestartet.
Im Folgenden kann der Ablauf der Rechnung über das Startfenster von Gabedit verfolgt werden. Dazu ist der Reiter mit der Dateiendung .log anzuwählen. Nach dem Start der Rechnung kann es einen Moment dauern, bis etwas in der im Output-Fenster angezeigt wird.


Der Verlauf der Rechnung kann über Klicken des Knopfes Update/end stückweise verfolgt werden. Eine erfolgreiche Rechnung wird mit dem Terminus EXECUTION OF GAMESS TERMINATED NORMALLY vermerkt. Sollten bei der Berechnung Probleme aufgetreten sein, sind an dieser Stelle mögliche Fehlerquellen vermerkt. Bei den Links befindet sich auch ein Beispieloutput mit dem groben Verlauf der generellen Informationen in einer Gamess log-File
Über den Knopf Geom. Conv. lassen sich die Iterationsschritte der Strukturoptimierung auch graphisch verfolgen, indem man den jeweiligen Punkt auf der Kurve im sich neu öffnenden Fenster auswählt und mit Draw bestätigt.


Im sich öffnenden Draw geometry-Fenster ist das O2-Molekül mit den berechneten Strukturparametern zu sehen. Vergleichen Sie den den berechneten Wert für die Bindungslänge mit dem Literaturwert!
Beim Klicken auf Dens. Orb. öffnet sich ein weiteres Fenster, in dem nun auch die berechneten Orbitale des Moleküls dargestellt werden können.

Um sich die Orbitale des O2-Moleküls anzeigen zu lassen, muss über einen Rechtsklick im Fenster unter der Rubrik Orbitals der Menüpunkt Read geometry and orbitals from Gamess output file angewählt werden. Im sich neu öffnenden Fenster ist die zuletzt erstellte Ausgabedatei bereits standardmäßig angewählt. Andernfalls wechseln Sie zum Verzeichnis, in dem sie die Eingabedatei gespeichert haben und wählen die zugehörige .log-File aus.

Nach Betätigen des Knopfes Open öffnet sich das Orbitals-Fenster.

Hier sind in der linken Spalte die Energien der berechneten Orbitale, deren Besetzungsgrad und Symmetrie angegeben. In der rechten Spalte finden Sie den Beitrag der jeweiligen Atomorbitalwellenfunktionen zur Wellenfunktion des links angewählten Molekülorbitals.
Um sich beispielsweise das berechnete HOMO anzeigen zu lassen, wählt man es in der linken Liste aus und klickt auf OK. Im sich neu öffneden Fenster Calculations of grid for an orbital wird festgelegt, in welchem Raumbereich und in welcher Pixeldichte die Orbitale gerendert werden sollen. Bei ungeeigneter Wahl des Minimum- und Maximumwerts kommt es zu "abgeschnittenen" Orbitalen. Sollte dies der Fall sein, verwenden Sie betragsmäßig größere Werte für das Minimum und Maximum.

Bei nochmaligem Bestätigen mit OK gelangt man zum Fenster Calculations of isosurfaces for an orbital. Der Isovalue gibt an, wie weitreichend das dargestellte Orbitallappenvolumen sein soll. Da klassischerweise ein Orbital als Volumen definiert ist, in dem sich das Elektron zu 99 % Wahrscheinlichkeit befindet, ist hier eine Funktion integriert, mit der man per Knopfdruck genau diesen Isovalue berechnen und anwenden lassen kann ("99 % Get Isovalue").

Nach Bestätigen mit OK wird nach kurzer Berechnungszeit im Fenster Orbitals/Density/Vibration das HOMO angezeigt. Dieses kann mit den bekannten Werkzeugen von allen Seiten betrachtet werden. Aus dem qualitativen MO-Schema für O2 erwartet man als HOMOs zwei einfach besetzte, energetisch entartete 2 π-Orbitale. Im Orbitals-Fenster ist dieser Umstand auch zu erkennen. Die "Alpha-Orbitale" 8 und 9 sind nicht zusätzlich mit einem beta-Spin-Elektron besetzt und weisen die selbe Energie von -0,581200 H auf.

Das Orbitals-Fenster lässt sich nun auch über Rechtsklick->Orbitals->Selection wieder aufrufen. Zudem kann eine Diaschau aller Orbitale über Rechtsklick->Orbitals->Slideshowerstellt werden.

Die Bezeichnung "Homo-8" ist wörtlich zu nehmen und bedeutet, dass dort das Orbital dargestellt ist, welches energetisch 8 Orbitale unter dem HOMO-Niveau liegt. Die Orbitalenergien können mit dem in Gabedit implementierten Einheitenumrechner in gängige Einheiten wie eV oder kJ × mol–1 umgerechnet werden.
- Gamess hat oft Probleme, die Symmetrie von heteroatomaren Molekülen korrekt zu verarbeiten und zu erkennen. Sollten Rechnungen nicht durchlaufen, versuchen Sie beispielsweise die Rechnung mit der festgelegten Punktgruppe C1 zu starten.
- Beim Bauen der Moleküle ist darauf zu achten, die Atomabstände nicht zu groß zu wählen, sondern in einem geeigneten Rahmen zu belassen. Sind die Atomabstände zu groß, werden lediglich die Atomorbitale der Einzelatome berechnet, liegen sie zu nahe beieinander wird die Rechnung abgebrochen.
- Soll beispielsweise die Punktgruppe von H2O von Gabedit korrekt als C2v erkannt werden, so müssen auch die Bindungslängen der H-O-Bindungen beim Erstellen des Moleküls auf dieselbe Länge gebracht werden. Dies kann entweder über die Messfunktion oder über die Rechtsklickfunktion Molecular mechanics->Optimization geschehen.
- Sollte eine Rechnung einfach kein Ende finden (Rechenzeit > 6 h), ist dies für die gegebenen Moleküle untypisch. Bis auf die Benzen-Aufgabe liegen die Rechenzeiten der anderen Moleküle im Minutenbereich. Starten Sie die Rechnung entweder mit denselben Parametern neu oder verändern sie manuell den Grenzwert wie bei Benzen gezeigt oder verwenden Sie einen anderen Basissatz.
- Wenn Sie manuelle Änderungen an der Eingabedatei vornehmen, speichern Sie diese zuerst ggf. unter einem neuen Dateinamen, bevor Sie die Rechnung starten.
- Bei manuellen Änderungen der Eingabedatei muss auf die korrekte Form geachtet werden. Auch Leerzeichen müssen richtig gesetzt sein, z.B. zwischen einem Parameter und $END.
- Die log-File wird in Windows als Textdatei deklariert, schauen Sie hier beim Suchen also nicht unbedingt auf die Dateiendung, sondern suchen Sie im jeweiligen Ordner nach einer Textdatei.
- Riggenmann, T., Einführung in die computerchemische Betrachtung basischemischer Sachverhalte mittels frei erhältlicher Software sowie Darstellung von {FeNO}7-Komplexen mit Citrato- und Tartratoliganden, Zulassungsarbeit im Fachbereich Anorganische Chemie, 2011
Einführung in die Grundbegriffe der Computerchemie, sowie beispielhafte Anwendung der Programmkombination Gabedit/Gamess.- http://www.msg.ameslab.gov/tutorials/tutorials.html
Material der Gamess-Bezugsseite.- http://sites.google.com/site/allouchear/Home/gabedit/gabedittutorialfiles/start
Beispiele für die Erstellung von Molekülen mit Gabedit und der Rechnung mit verschiedenen Programmen, darunter auch Gamess.- http://sites.google.com/site/allouchear/Home/gabedit/manual
Das Handbuch für Gabedit.- http://www.msg.ameslab.gov/gamess/documentation.html
Das Handbuch für Gamess.- http://muqchem.millersville.edu/raw%20output.pdf
Beschreibt grob den Inhalt einer Gamess log-File
Auch im Rahmen des schulischen Chemieunterrichts lässt sich die Programmkombination Gabedit/Gamess gezielt einsetzen. Nicht nur in einem Semniarteil der gymnasialen Oberstufe ist der Gebrauch möglich, sondern auch schon in unteren Klassen, beispielsweise zur eigenständigen Berechnung von Tendenzen der Ionisierungsenergie, Bindungslänge und Elektronenaffinität und bietet neben dem praktisch-chemischen Arbeiten eine weitere Alternative, chemische Fachinhalte erfahrbar zu machen. Zudem können die Ergebnisse solcher Berechnungen von den Schülerinnen und Schülern auf unteschiedlichste Weise aufgearbeitet und dargestellt werden und damit die Medienkompetenz der Schülerinnen und Schüler auch im Fach Chemie zu fordern und zu fördern.
Sollten Sie sich weitergehend mit schulischen Anwendungsmöglichkeiten solcher Programme beschäftigen wollen, können Sie sich gerne am Aufbau einer Internetseite beteiligen. Ihr Ansprechpartner ist Tobias Riggenmann (Kontaktdaten über P. Mayer).
Für jede Rechnung muss ein Protokoll erstellt werden, das grundsätzlich folgende Elemente enthält:
- Kopfzeile mit den üblichen Angaben (Name des Praktikanten, Einrichtung die das Praktikum ausrichtet, Bezeichnung des Praktikums, Versuchsnummer, Datum)
- Überschrift mit Versuchsbezeichnung
- Einleitung (Beschreibung des Versuchsgegenstandes und des Versuchsziels in ein bis zwei Sätzen)
- Material und Methoden (Verwendete Programme mit Versionsnummer, Rechenparameter, Leistungsdaten des verwendeten Computers)
- Ergebnisse
- Diskussion
- Literatur- und Quellenangaben
- Anhang mit Daten der Eingabedatei (z.B. als Screenshot)
Solle es Probleme mit den Programmen oder bei der Berechnung geben, speichern Sie die .log-Datei sowie die .inp-Datei und wenden Sie sich an Ihre Assistenten. Versuchen Sie auch mit den Rechenparametern zu spielen, sollte eine Rechnung nicht ordnungsgemäß durchlaufen (das können Sie übrigens nur mit Sinn und Verstand, wenn sie sich über die Grundbegriffe informiert haben!).
Die Rechenparameter für die Rechnungen sind in folgendem Format wiedergegben:
Symmetry; Run type; SCF type; Max # Iterations; Correlation type; Correlation method; (Localized type;) Basis set; #d heavy atom polarisation functions; #f heavy atom polarisation functions; #light atom polarisation functions; Polar;
Als Ligand bindet CO in Übergangsmetallkomplexen stets mit seinem C-Atom an das Zentralmetall. Betrachten Sie die Form der
MOs und deren Energie. Welches MO hat die geeignete Symmetrie für die σ-Donor-Bindung und von welchem Atom geht sie aus?
Als Ligand stellt CO auch MOs bereit zur π-Rückbindung von Metall-d-Orbitalen zum Ligand. Welche CO-Orbitale haben die
geeignete Symmetrie?
Schauen Sie sich die mesomeren Grenzstrukturen der Lewis-Formel von CO an. Welche Bindungsordnung erwarten Sie? In der Literatur
wird für den Bindungsabstand
ein Wert von 112,8 pm (oder 1,128 Angstrom) angegeben. Welchen Wert ergibt die Rechnung (log-File, oder Messfunktion). Welcher Bindungsordnung entspricht dieser Abstand? Zur Beantwortung dieser
Frage können Sie in Lehrbüchern nach Bindungsabständen für C-O-Bindungen verschiedener Bindungsordnungen schauen.
Die Bindungsordnung hat auch entscheidenden Einfluss auf die Schwingungsfrequenz der betreffenden Bindung.
In der Literatur findet man für die Schwingungsfrequenz von freiem CO einen Wert von 2143 cm–1. Welchen
Wert erhalten Sie aus dieser Rechnung? Hierfür müssen Sie zuerst die Struktur optimieren und anschließend die berechneten Strukturparameter in einer neuen Rechnung für die Schwinungsfrequenzen
benutzen. Verwenden Sie dazu die Rechtsklickfunktion Geometry->Read last geometry from Gamess output file. Anmerkung: Die berechneten Werte sind in aller Regel zu groß Sie dürfen die berechneten
Werte ggf. mit 0,95 multiplizieren. Sie finden den Wert entweder im log-File, das Sie außer in Gabedit auch in einem ganz normalen Texteditor
öffnen können oder im Orbitals/Density/Vibrations-Fenster, wenn Sie den entsprechenden log-File laden und anschließend per Rechtsklick unter Animation->Vibration->File->Read->Read a Gamess output file die einzelnen Schwingungen anzeigen lassen und
unter Tools auch das zugehörige IR-Spektrum zeichnen lassen.
CO siedet bei –191,55 °C, Stickstoff bei –195,79 °C. Die Siedepunkte sind erstaunlich ähnlich, obwohl man
für CO einen deutlich höheren Siedepunkt erwarten würde, oder? Warum sind die Siedepunkte so ähnlich? Für freies CO wird in der
Literatur ein Dipolmoment von 0,11 Debye angegeben. Was ergibt die Rechnung (suchen Sie im log-File nach dem Dipolmoment)?
Worauf beruht das kleine Dipolmoment von CO?
Rechenparameter
Für die Strukturoptimierung: fixed C1; Equilibrium geometry; RHF; 90; MP2; MP2; 6-331G; 2; 0; 1; default
Für die Frequenzberechnung: fixed C1; Frequencies; RHF; 90; MP2; MP2; 6-331G; 2; 0; 1; default
Sie müssen wieder zuerst die Struktur optimieren und anschließend die berechneten Strukturparameter in einer neuen Rechnung für die Schwinungsfrequenzen benutzen. Verwenden Sie dazu die Rechtsklickfunktion Geometry->Read last geometry from Gamess output file. Betrachten Sie die Schwingungen des berechneten Moleküls. Welche Schwingungen sind IR-aktiv und welche Frequenzen haben die Schwingungen? Nun betrachten Sie den Wert für die asymmetrische Valenzschwingung: weshalb ist dieser größer als die Schwingungsfrequenz in CO, die Sie im vorigen Beispiel berechnet haben? Bedenken Sie, dass in CO eine Dreifachbindung als wichtigste Grenzstruktur formulierbar ist! Vergleichen Sie auch mit dem Literaturwert der IR-Schwingungen von CO2.
Rechenparameter
Für die Strukturoptimierung: fixed C1; Equilibrium geometry; RHF; 90; MP2; MP2; 6-331G; 2; 0; 1; default
Für die Frequenzberechnung: fixed C1; Frequencies; RHF; 90; MP2; MP2; 6-331G; 2; 0; 1; default
Beachten Sie, dass NO ein Radikal ist und deshalb die Rechenmethode unrestricted sein muss. Welches Dipolmoment und welcher Bindungsabstand werden berechnet. Vergleichen Sie mit den Werten für CO. Entsprechen
die Ergebnisse Ihren Erwartungen?
Um zu erfahren, wo das ungepaarte Elektron lokalisiert ist, können Sie eine NBO-Rechnung durchführen. Visualisieren Sie wieder die berechneten Orbitale. Sie stellen fest, dass sich die Form der Orbitale im Vergleich
zur MP2-Rechnung geändert hat.
An welchem Atom vermuten Sie das ungepaarte Elektron? Von welcher Art ist das Orbital, in dem sich das ungepaarte Elektron
befindet? Diese Frage können Sie auch beantworten, wenn Sie die normalen MOs betrachten.
NO dimerisiert zu N2O2. Wie wird die Lewis-Formel des Dimers aussehen? Wenn Sie mögen, können Sie
die Rechnungen für das richtige Dimer und für ein falsches Dimer (z.B. ONON) durchführen und vergleichen.
NO (genauer NO+) agiert ähnlich wie CO als Ligand in Komplexverbindungen. Gute σ-Donorwirkung ist dann möglich, wenn die Orbitallappen möglichst
ausladend sind und gut mit den Metallorbitalen überlappen können. Welches Atom in NO hat in dieser Hinsicht bessere Voraussetzungen?
Wie sind die Unterschiede im Vergleich zu CO? Beachten Sie, dass Sie ein Elektronenpaar brauchen, d.h. sie benötigen zwei
Elektronen, je eines mit alpha- und beta-Spin, die sich in Orbitalen ähnlichen Aussehens und ähnlicher Energie befinden.
Rechenparameter
Für die Strukturoptimierung mit normalen MOs: fixed C1; Equilibrium geometry; UHF; 90; MP2; MP2; 6-331G; 2; 0; 1; default; Ergänzen von projct=.f. im $STATPT-Block der Eingabedatei (Danach speichern und dann erst berechnen lassen!)
Für die Berechnung von NBOs: fixed C1; Equilibrium geometry; UHF; 90; Localized Type=Pipek-Mezey; 6-331G; 2; 0; 1; default; Ergänzen von projct=.f. im $STATPT-Block der Eingabedatei (Danach speichern und dann erst berechnen lassen!)

Der zusätzlich eingefügte Befehl bedingt, dass die Rotationsfreiheitsgrade der Atome bei der Berechnung der Struktur außen vor gelassen werden.
Beachten Sie, dass auch NO2 ein Radikal ist. Welchen Bindungswinkel erhalten Sie aus der Rechnung? Vergleichen Sie mit dem Literaturwert.
Führen Sie dann eine NBO-Rechnung durch. An welchem Atom vermuten Sie das ungepaarte Elektron? Von
welcher Art ist das Orbital, in dem sich das ungepaarte Elektron befindet?
NO2 dimerisiert zu N2O4. Wie wird die Lewis-Formel des Dimers aussehen? Welche Symmetrie (Punktgruppe)
hat das Molekül? Freiwillig: Rechnen Sie N2O4. Entspricht die Symmetrie des Moleküls Ihren Erwartungen?
Rechenparameter
Für die Strukturoptimierung von NO2mit normalen MOs: Detected as C2v; Equilibrium geometry; UHF; 90; MP2; MP2; 6-331G; 2; 0; 1; default;
Für die Berechnung der NBOs von NO2: Detected as C2v; Equilibrium geometry; UHF; 90; Localized Type=Pipek-Mezey; 6-331G; 2; 0; 1; default;
Benzen wird auch in Schulbüchern MO-theoretisch besprochen. In welchen Orbitalen sind die sechs π-Elektronen untergebracht? Lassen Sie sich von den Energien der Orbitale nicht in die Irre führen, nach dieser Rechnung sind die π-Orbitale nicht die energetisch höchsten besetzten MOs. Die Symmetrie bzw. die Art der Orbitale ist entscheidend.
Sollten die gegebenen Parameter zu einer "unendlich langen Rechnung" (> 6 h Rechenzeit) ohne Erreichen des Grenzwertes ("OptTol") werden, verwenden Sie einen kleineren Basissatz oder setzen Sie den Grenzwert in der Eingabedatei manuell auf einen höheren Wert, beispielsweise 1 × 10–4. Vermerken Sie solche Änderungen im Protokoll!

Mit den nachfolgenden Paramtern dauerte die normale MO-Rechnung knapp viereinhalb Stunden, die NBO-Rechnung 70 Minuten (Acer Aspire 5920G, 2 GHz Intel Core 2 Duo Prozessor, 2 GB DDR-RAM, Windows 7 Professional 32-bit).
Rechenparameter
Für die Strukturoptimierung mit normalen MOs: fixed C1; Equilibrium geometry; UHF; 90; MP2; MP2; 6-331G; 2; 0; 1; default; Ergänzen von projct=.f. im $STATPT-Block der Eingabedatei; Ändern der OptTol auf 1e–4 (Danach speichern und dann erst berechnen lassen!);
Für die Berechnung von NBOs: fixed C1; Equilibrium geometry; UHF; 90; Localized Type=Pipek-Mezey; 6-331G; 2; 0; 1; default; Ergänzen von projct=.f. im $STATPT-Block der Eingabedatei; Ändern der OptTol auf 1e–4 (Danach speichern und dann erst berechnen lassen!)
...spielen Sie mit der Software! Sollten Sie Gefallen am Rechnen gefunden haben, versuchen Sie doch diese Rechnungen mit anderen Parametern, beispielsweise anderen Basissätzen oder mal per DFT und vergleichen Sie die Ergebnisse. Vielleicht reizt es Sie auch, die jeweiligen Parameter für die optimale Reproduktion der Literaturwerte von Bindungslängen und -winkeln zu finden!
Tobias Riggenmann
Literatur: D. R. Palleros, Experimental Organic Chemistry, Wiley, New York, 2000, Seite 645--652. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek: VK 5100 P166. Originalliteratur: P. Besse, J. Bolte, H. Veschambre, J. Chem. Ed. 1995, 72, 277--278; E. C. S. Brenelli, P. J. S. Moran, J. A. R. Rodrigues, Synth. Comm. 1990, 20, 261--266; R. Chenevert, S. Thiboutot, Chem. Lett. 1988, 1191--1192.
Bäckerhefe bitte selbst mitbringen!
Benötigte Chemikalien: trockene Bäckerhefe, Wasser, 1-Phenyl-1,2-propandion, tert-Butylmethylether, wasserfreies Magnesiumsulfat, Cyclohexan, DC-Platten mit Silica Gel, Deuterochloroform für NMR.
10 g trockene Bäckerhefe werden in einem 250-mL Erlenmeyerkolben in 35 mL Wasser gerührt bis eine gleichmäßige Mischung vorliegt. Dann fügt man 0,23 mL 1-Phenyl-1,2-propandion hinzu und rührt bei Raumtemperatur. Den Verlauf der Reaktion können Sie verfolgen, indem Sie zum Zeitpunkt des Reaktionsbeginns sowie darauf alle 15 Minuten mit einer Pipette ungefähr 0,2 mL der Reaktionsmischung entnehmen und mit 0,4 mL tert-Butylmethylether in einem kleinen Präparateglas ausschütteln, einen Tropfen auf einer Silica Gel Platte anbringen und zum Reaktionsende nach frühestens 90 Minuten untersuchen (Laufmittel Cyclohexan/tert-Butylmethylether im Verhältnis 6:4). Am Ende der Umsetzung füllen Sie die Hefemischung in einen Scheidetrichter und geben 50 mL tert-Butylmethylether hinzu und schüttlen vorsichtig aus (nicht zu stark, da sonst keine saubere Phasengrenze erhalten wird; von Zeit zu Zeit belüften nicht vergessen!). Nach 5 Minuten lassen Sie die Phasen trennen, geben die untere wässrige Phase in den Erlenmeyerkolben der Umsetzung, die organische Phase in einen separaten Erlenmeyerkolben. Schütteln Sie die wässrige Phase nochmals mit 35 mL tert-Butylmethylether aus und fügen Sie die organische Phase der anderen zu. Die wässrige Phase schütteln Sie dann ein drittes Mal aus mit 35 mL tert-Butylmethylether und vereinigen die organischen Phasen wieder. Geben Sie etwa 4 g wasserfreies Magnesiumsulfat zur organischen Phase und lassen Sie sie wenigstens 10 Minuten stehen. Dann filtrieren Sie das Magnesiumsulfat ab und lassen den Ether langsam verdampfen (kann über Nacht in einer Abdampf- oder Kristallisierschale geschehen oder am Rotavapor). Dann lassen Sie noch eine Weile einen leichten Stickstoffstrom durch das erhaltene Öl blasen um letzte Lösungsmittelspuren zu beseitigen.
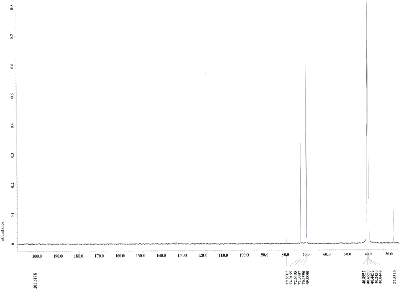
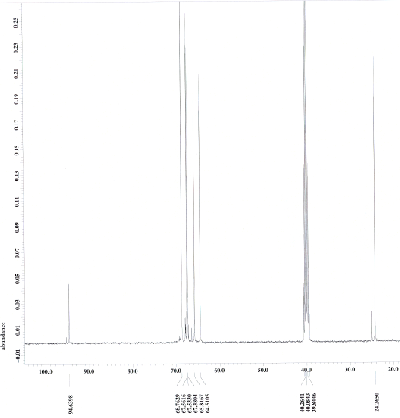
Nehmen Sie vom Reduktionsprodukt ein 1H-NMR-Spektrum sowie ein 13C-NMR-Spektrum aus CDCl3 auf und interpretieren Sie das Spektrum. Zum Vergleich sehen Sie das 1H-NMR-Spektrum und das 13C-NMR-Spektrum jeweils aus CDCl3 des Edukts unten abgebildet.
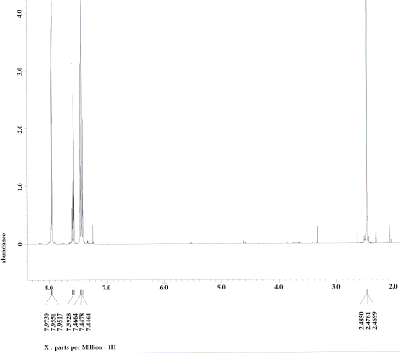
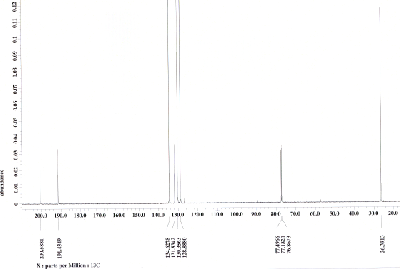
Nehmen Sie vom Reduktionsprodukt ein IR-Spektrum auf und interpretieren Sie das Spektrum. Zum Vergleich sehen Sie das IR-Spektrum des Edukts unten abgebildet.
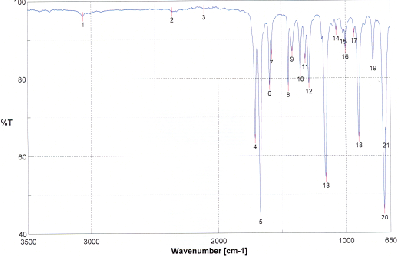
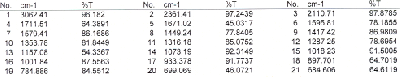
Welche Produkte sind bei der Reaktion theoretisch möglich? Erläutern Sie am Beispiel des bei der Reaktion wirklich entstehenden Stereoisomers, welche Konformere begünstigt sind und weshalb sie dies sind. Welchen Einfluss hat die unterschiedliche Stabilität der Konformere auf die Kopplungskonstante zwischen den beiden C-gebundenen H-Atomen an den Hydroxyl-C-Atomen? Versuchen Sie diese Kopplungskonstante aus dem NMR-Spektrum des Produkts abzuleiten.
Literatur: J. D. Woollins in Inorganic Experiments, Wiley VCH, Weinheim, 2003, Seite 85--87. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek: VH 5100 W913(2).
Benötigte Chemikalien: Dicyclopentadien, Eis, Diethylether, Kaliumhydroxid (Flocken), Eisen(II)-chlorid Tetrahydrat, Dimethylsulfoxid, verdünnte Salzsäure, Wasser, Essigsäureanhydrid, Ortophosphorsäure, Calciumchlorid zum Trocknen, Natriumbicarbonat, Natrium-tetrahydroborat, Diethylether, wasserfreies Magnesiumsulfat, 40/60 Petrolether.
Darstellung von Cyclopentadien
Cyclopentadien dimerisiert bei Raumtemperatur sehr leicht und muss daher erst frisch destilliert werden. Setzen Sie wenigstens 50 mL Dicyclopentadien ein. Es werden jedoch nur etwa 4,3 mL Cyclopentadien benötigt. Cyclopentadien destilliert bei ca. 42--44 °C über. Damit die Retro-Diels-Alder-Reaktion überhaupt ablaufen kann, wird eine Ölbadtemperatur von 180 °C benötigt. Den Destillatkolben gut im Eisbad kühlen!
Darstellung von Ferrocen
Während Cyclopentadien destilliert wird, bauen Sie einen 250-mL-Zweihalskolben versehen mit einem Rückflusskühler in der Mitte und einem 100-mL-Tropftrichter auf der Seite auf. Auf dem Rückflusskühler wird ein Blubberer mit einem zusätzlichen Hahn zum Anschluss von Stickstoff angebracht. 50 mL Diethylether und 20 g Kaliumhydroxid (in Schuppen) werden in der zuvor gründlich mit Stickstoff gespülten Apparatur gut gerührt. Während dessen löst man in einem mit einem Stopfen verschlossenen Rundkolben 5 g feingepulvertes Eisen(II)-chlorid Tetrahydrat in 20 mL entgastem Dimethylsulfoxid. Hierzu muss man etwa 1 Stunde kräftig rühren. Dann gibt man unter kräftigem Rühren und einem langsamen Stickstoffstrom 4,3 mL frisch detilliertes Cyclopentadien zu der KOH/Ether-Mischung. Nach 15 Minuten Rühren gibt man über den Tropftrichter die Eisen(II)-Lösung tropfenweise zur cyclopentadienhaltigen Lösung. Diese Reaktion ist exotherm und die Lösung kann beginnen zu kochen. Nach Beendigung der Zugabe der Eisen(II)-Lösung rührt man noch eine Stunde. Dann dekantiert man die Etherschicht ab, wäscht den Kolben mit weiteren 25 mL Ether und vereinigt die Etherphasen. Dann werden die Etherphasen zweimal mit je 20 mL verdünnter Salzsäure gewaschen, dann zweimal mit je 20 mL Wasser. Darauf wird der Ether vorsichtig am Rotavapor abgezogen und man erhält einen orangefarbenen Niederschlag von Ferrocen.
Darstellung von Acetylferrocen
In einem 50-mL-Rundkolben werden 10 mL Essigsäureanhydrid vorgelegt, 3 g Ferrocen hinzugegeben und anschließend unter Rühren tropfenweise mit 2 mL Orthophosphorsäure versetzt. Dann wird ein mit CaCl2 gefülltes Trockenrohr aufgesetzt. Man erhitzt dann die Mischung 20 Minuten lang auf einem Ölbad auf 110 °C und gießt dann die heiße Mischung auf 80 g gecrunchtes Eis. Man wäscht den Kolben mit etwas Eis aus und vereinigt die Eisphasen. Sobald das Eis geschmolzen ist, wird die Lösung mit festem Natriumhydrogencarbonat neutralisiert. Dann wird die Mischung 20 Minuten lang in Eis abgekühlt und dann der braune Feststoff abfiltriert. Der Feststoff wird im Vakuum getrocknet.
Reduktion von Acetylferrocen
Man löst 1 g Acetylferrocen in 15 mL Ethanol und gibt dann 5 mL Wasser hinzu. 0,8 g Natrium-tetrahydroborat werden in 4 mL Wasser gelöst und dann langsam unter Rühren zur Acetylferrocenlösung getropft. Dann lässt man die 15 Minuten lang stehen, wobei sie inzwischen hellgelb geworden sein sollte. Oft bleibt die Lösung jedoch braun. Um die Reduktion zu vervollständigen, gibt man noch etwas Natrium-tetrahydroborat hinzu. Dann fügt man 100 mL Wasser hinzu und extrahiert die wässrige Mischung zweimal mit je 20 mL Diethylether. Die Etherextrakte werden vereinigt und über Magnesiumsulfat getrocknet. Dann wird das Magnesiumsulfat abfiltriert und der Ether im Filtrat langsam am Rotavapor abgezogen. Das Produkt ist häufig ölig und kann aus 40/60 Petrolether umkristallisiert werden. Der Feststoff wird dann im Exsikator getrocknet.
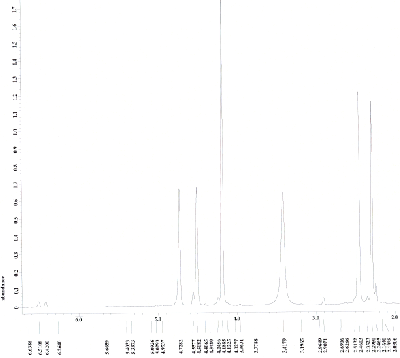
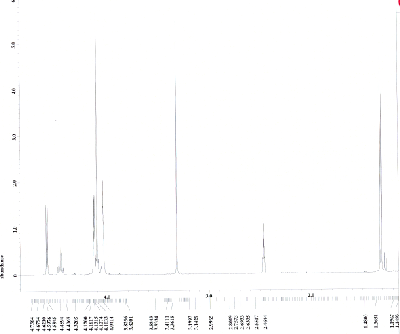
Nehmen Sie von Acetylferrocen und vom Reduktionsprodukt jeweils ein 13C-NMR-Spektrum aus deuteriertem Dimethylsulfoxid auf und interpretieren Sie die Spektren, welche den folgenden beiden ähneln sollten (Acetylferrocen oben, Reduktionsprodukt unten).
Die 1H-NMR-Spektren aus deuteriertem Dimethylsulfoxid sollten den folgenden ähneln (Acetylferrocen oben, Reduktionsprodukt unten).
Wie könnte ein plausibler Mechanismus für die Acetylierung aussehen? Berücksichtigen Sie, dass Ferrocen etwa 3 × 106 mal schneller reagiert als Benzol.
Literatur: A. D. Adler, F. R. Longo, J. D. Pinarelli, J. Goldmacher, J. Assour, L. Korsakoff, J. Org. Chem. 1967, 32, 476--476.
Vorsicht: Pyrrol ist giftig! Nur im Abzug arbeiten und Schutzhandschuhe tragen!
Benötigte Chemikalien: frisch destilliertes Pyrrol, Benzaldehyd, Propionsäure, Methanol (zum Waschen), Wasser.
Zunächst wird Pyrrol bei Normaldruck frisch destilliert. Pyrrol destilliert dabei farblos über. Die Temperatur des Ölbads kann
auf 150 °C gesetzt werden. Etwas Vorlauf wird verworfen. Dann werden 300 mL Propionsäure in einem 500 mL Zwei- oder
Dreihalskolben unter Rückfluss zum Sieden erhitzt (Temperatur des Ölbads auf 165 °C setzen). Zu der siedenden
Propionsäure werden dann 5,6 mL frisch destilliertes Pyrrol und 8,0 mL Benzaldehyd gegeben. Diese Mischung wird nach der Zugabe
dann noch 30 Minuten (nicht länger, da sonst die Ausbeute sinkt!) unter Rückfluss weitergekocht. Danach entfernt man das
Ölbad und lässt
die Mischung auf Raumtemperatur abkühlen. Dann wird abfiltriert und der Niederschlag erst gründlich mit Methanol gewaschen
und danach mit heißem Wasser. Dann lässt man den Niederschlag erst an Luft etwas trocknen, ehe er dann im Vakuum getrocknet
wird.
Überprüfen Sie dann die Reinheit des Produkts mittels eines UV-Spektrums. Lösen Sie dazu sehr wenig des Produkts in Dichlormethan, so dass eine schwach rotviolette Lösung entsteht. Füllen Sie diese in die Küvette, nachdem Sie zuvor die Baseline mit reinem Dichlormethan gemessen haben. Messen Sie den Bereich von 450--700 nm. Das Spektrum sollte dem folgenden ähneln.
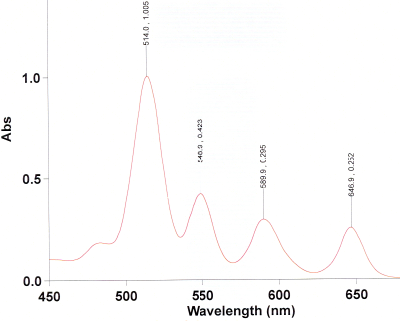
Nur, wenn die Bande bei 650 nm stärker ist als die bei 590 nm und 550 nm, oder gar stärker als die bei 515 nm, enthält ihr Produkt viel Tetraphenylchlorin. Dieses muss dann gemäß den Angaben in K. Rousseau, D. Dolphin, Tetrahedron Lett. 1974, 15, 4251--4254 dehydriert werden. Wenn das UV-Spektrum jedoch so aussieht wie das obige, dann ist der folgende Schritt nicht nötig!
Dehydrierung von Tetraphenylchlorin
1,0 g des verunreinigten Tetraphenylporphins werden in 500 mL Toluol gelöst und unter Rückfluss gekocht. Dazu wird 0,25 g 2,3-Dichloro-5,6-dicyanobenzochinon gegeben und noch ein halbe Stunde weitergekocht. Danach wird auf Raumtemperatur abgekühlt und mit 500 mL einer 1%igen Natriumhydroxidlösung, die 0,5 g Natriumdithionit enthält, ausgeschüttelt. Die organische Phase wird dreimal mit je 100 mL Wasser gewaschen und anschließend mit Natriumsulfat getrocknet. Dann wird das Toluol am Rotavapor abgezogen und der Niederschlag wird aus Dichlormethan/Methanol umkristallisiert.
Anmerkung für die Assistenten: Die sich als rein erweisenden Fraktionen aller Versuchsgruppen werden in einem einzigen Präparateglas oder Kolben gesammelt. Die verunreinigten Fraktionen werden sachgerecht entsorgt.
Synthese von [5,10,15,20-Tetraphenyl-21H,23H-porphinato(2–)]-Nickel(II)
Literatur: E. C. Johnson, D. Dolphin, in Inorg. Synth. 20, 143--153. Standortsignatur der Fachbibliothek: VH 5500.
Benötigte Chemikalien: Tetraphenylporphin, Trifluoressigsäure, Eisessig, Nickel(II)-acetat Tetrahydrat, verdünnte Essigsäure, Methanol, Wasser.
0,5 g trockenes Tetraphenylporphin werden in möglichst wenig Trifluoressigsäure gelöst (etwa 7,5 mL). Diese Lösung wird zu 100 mL unter Rückfluss kochendem Eisessig gegeben. Dann löst man 1,0 g Nickel(II)-acetat Tetrahydrat in möglichst wenig heißer verdünnter Essigsäure (ca. 5 mL) und tropft diese Lösung langsam innerhalb von 5 Minuten zu der unter Rückfluss kochenden Eisessiglösung. Nach beendetem Zutropfen wird die Mischung dann noch eine Stunde unter Rückfluss gekocht. Danach wird heiß abfiltriert und der Niederschlag so lange mit kochender Essigsäure gewaschen, bis das Filtrat farblos ist. Danach wird noch dreimal mit je etwa 12 mL Wasser gewaschen, dann dreimal mit je etwa 12 mL Methanol. Das rotviolette kristalline Produkt wird dann im Vakuum getrocknet.
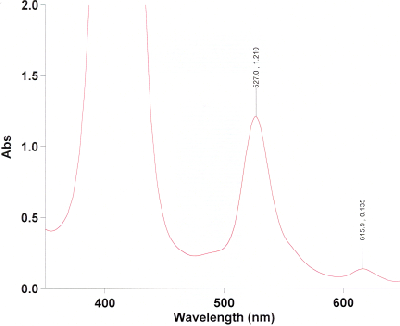
Nehmen Sie vom Produkt ein UV-Spektrum aus Methylenchlorid auf und interpretieren Sie das Spektrum, welches dem folgenden ähneln sollte. Vergleichen Sie es mit dem UV-Spektrum des reinen Liganden.
Literatur: C. Husson, L. Odier, P. J. A. Vottero,
Carbohydr. Res. 1998, 307, 163--165;
H. G. Becker, W. Berger, G. Domschke, Organikum, New York, 20. Auflage 1999, S. 703 f.
Vorsicht: Bitte beachten Sie die fachgerechte Entsorgung von Raney-Nickel! Stäube von Nickel und Nickelverbindungen können Krebs auslösen!! Abzug!! Die Isomerisierung von myo-Inosit muss im Nachtlabor durchgeführt werden.
Benötigte Chemikalien: myo-Inosit, Nickel-Aluminium-Legierung (gepulvert), Natriumhydroxid, Wasser, Ethanol. Zur Entsorgung von Raney-Nickel werden benötigt: konz. Salzsäure, 30%iges Wasserstoffperoxid, Natriumcarbonat.
Darstellung von frischem Raney-Nickel
In einem 1000 mL Becherglas werden 15 g einer ca. 50% Nickel enthaltenden gepulverten Nickel-Aluminium-Legierung
mit 150 mL Wasser aufgeschwemmt. Dazu gibt man ohne Kühlen vorsichtig ca. 25 g festes Natriumhydroxid. Die heftig siedende
Mischung soll dabei gerade nicht überschäumen. Nach Zugabe des Natriumhydroxids wird ca. 15 Minuten bei Raumtemperatur
stehen gelassen und anschließend für 30 Minuten auf 70 °C warm gehalten und dabei gelegentlich geschüttelt (keinen
Rührer bzw. Rührfisch verwenden!!).
Es setzt sich ein schwammiger Niederschlag ab. Die überstehende, trübe Lösung wird vorsichtig dekantiert. Der Niederschlag wird
fünfmal mit 50 mL Wasser aufgeschwemmt, geschüttelt und nach Absetzen des Niederschlags dekantiert.
Isomerisierung von myo-Inosit
In einem 250 mL Rundkolben werden 10 g myo-Inosit unter Rühren und mäßigem Erwärmen in einem Ölbad in 25 mL Wasser
gelöst. Dazu gibt man unter Rühren sämtliches nach obiger Vorschrift frisch hergestelltes Raney-Nickel. Man erhitzt zum Sieden
und lässt für ca. 24 Stunden unter Rückfluss und Rühren kochen.
Aus der Reaktionsmischung wird anschließend durch Filtration mit einem Glasfiltertiegel das Raney-Nickel entfernt und mit ca.
25 mL Wasser gewaschen. Das Waschwasser wird dem Filtrat zugefügt. Beachten Sie die weiter unten angegebene Vorschrift zur
Entsorgung von Raney-Nickel!
Aufbereitung und Reinigung
Die rötliche Lösung wird am Rotationsverdampfer bei 80 °C auf etwa 10--20% eingeengt. Sobald ein Feststoff auszufallen beginnt, wird nicht mehr weiter eingeengt. Zur warmen Lösung werden ca. 40 mL siedendes Ethanol gegeben und der Niederschlag abgenutscht und mit ca. 30 mL siedendem Ethanol gewaschen. Die Filtrate werden vereinigt und zur späteren Aufbereitung gesammelt! Der gewaschene Niederschlag (ca. 1 g) wird in wenig siedendem Wasser (ca. 11 mL) gelöst, auf Raumtemperatur abgekühlt und über Nacht in den Kühlschrank gestellt. Am folgenden Praktikumstag wird das Produkt abgenutscht. Die Mutterlauge lässt man langsam an Luft einengen, worauf weiteres Produkt anfällt.
Entsorgung von Raney-Nickel (FU Berlin, OC Grundpraktikum)
Für 100 g des Katalysators gilt folgende Vorschrift (für kleinere Mengen entsprechend skalieren):
Im Abzug wird in einem ausreichend großen Becherglas (2000 mL für 100 g Katalysator) der Katalysator mit 25%iger Salzsäure
übergossen und die Mischung erwärmt. Das Nickel geht dabei allmählich in Lösung. Durch vorsichtige Zugabe von 30%igem
Wasserstoffperoxid kann der Auflösungsvorgang beschleunigt werden, wobei die Dosierung vorsichtig erfolgen muss. Von Zeit zu
Zeit wird Säure hinzugegeben, so dass die Reaktion aufrechterhalten bleibt und die sich bildenden Nickelsalze keine gallertartigen
Koagulate bilden. Für 100 g Katalysator benötigt man etwa 400 mL Säure. Die Reaktion ist zu Ende, wenn der grauschwarze
Bodensatz des Katalysators verschwunden ist und eine klare dunkelgrüne Lösung vorliegt. Diese Lösung wird bis zum Ausbleiben
von Aufschäumen mit konzentrierter Natriumcarbonatlösung versetzt. Der gallertartige Niederschlag besteht aus basischem
Nickelcarbonat, welches abfiltriert und feucht in die Feststofftonne gegeben wird. Das Filtrat kann ins Abwasser gegeben
werden, wenn es farblos ist. Ansonsten stehenlassen, Nachfällung abwarten, erneut filtrieren.
Nehmen Sie vom Produkt ein 13C-NMR Spektrum aus D2O auf und interpretieren Sie das Spektrum. Der folgenden Abbildung können Sie entnehmen, dass die Umsetzung mit Raney-Nickel zu einem Gemisch von Isomeren führt, die allesamt in der Mutterlauge nachzuweisen sind. Erst durch die Fällung mit Ethanol wird scyllo-Inosit angereichert. Durch Vergleich ihres Spektrums mit den unten abgebildeten können Sie auf den Reinheitsgrad ihrer Verbindung schließen.
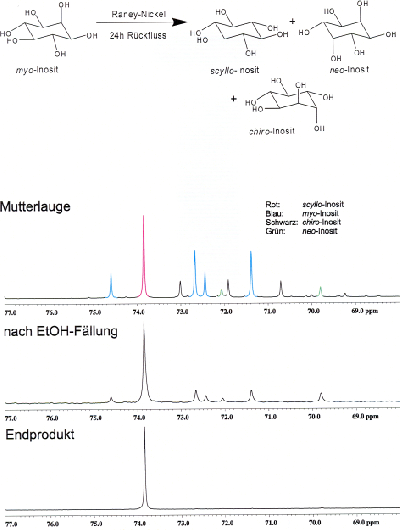
Literatur: M. Schröder in Inorg. Experiments, Wiley VCH, Weinheim, 2003, Seite 168--170. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek: VH 5100 W913(2).
Vorsicht: Nickelsalze sind potentiell krebserregend. Hautkontakt vermeiden!
Benötigte Chemikalien: 1,2-Diaminoethan, Methanol, konz. Bromwasserstoffsäure, Diethylether zum Waschen, Aceton, Wasser, Nickel(II)-carbonat basisch, Natriumperchlorat.
Darstellung von 1,2-Diaminoethan Dihydrobromid
2,0 g 1,2-Diaminoethan werden in 7 mL Methanol gelöst. Dann wird die Lösung in einem Eisbad gekühlt und vorsichtig 12 mL konz. HBr tropfenweise und unter Rühren zugegeben. Der Niederschlag wird abfiltriert, mit Diethylether gewaschen und im Vakuum getrocknet bis zur Gewichtskonstanz (immer wieder Gewicht von Kolben und Substanz messen). Durch Einengen des Filtrats oder durch Zugabe von Diethylether zum Filtrat kann die Ausbeute gesteigert werden.
Darstellung des Makrocyklus
Eine Mischung aus 11,1 g 1,2-Diaminoethan Dihydrobromid, 100 mL Aceton und 3,0 g 1,2-Diaminoethan wird 30 Minuten unter Rückfluss erhitzt. Dann lässt man abkühlen, filtriert den Niederschlag ab, und wäscht ihn zuerst mit eiskaltem Aceton, dann mit Diethylether und trocknet ihn daraufhin im Vakuum bis zur Gewichtskonstanz.
Darstellung des Nickel(II)-komplexes
5,0 g des zuvor hergestellten Makrocyklus werden in möglichst wenig Wasser gelöst (etwa 25 mL) und mit einem leichten Überschuss an Nickel(II)-basisch-carbonat (1,5 g) versetzt (ca. 20% Überschuss an Nickel(II); bitte beachten, wenn technische Nickel(II)- carbonat eingesetzt wird!). Die Mischung wird ca. 30 Minuten auf etwa 70 °C erhitzt. Danach wird abfiltriert und das Filtrat mit 3 mL einer gesättigten wässrigen Natriumperchloratlösung versetzt. Die Lösung wird dann auf einem Eisbad abgekühlt, worauf sich ein gelber, kristalliner Niederschlag bildet.
Das Produkt ist ein Diastereoisomerengemisch. Das reine N-meso Diastereoisomer kann durch schnelle Umkristallisation aus heißem Methanol gewonnen werden.
Analog zur Synthese des Nickel(II)-komplexes kann ein Kupfer(II)-komplex dargestellt werden, wenn anstelle von Nickel(II)- carbonat Kupfer(II)-carbonat, basisch, eingesetzt wird. Das N-meso Diastereoisomer ist orange, das N-racemische Diastereoisomer ist rot. Das N-meso Diastereoisomer kann durch Umkristallisation aus Wasser/Ethanol gewonnen werden, das N-racemische Diastereoisomer durch Extraktion in kochendes Dioxan.
Nehmen Sie vom reinen Makrocyklus ein IR-Spektrum auf, das dem folgenden entsprechen sollte. Identifizieren Sie die charakteristische C=N Streckschwingung.
Nehmen Sie vom Nickel(II)/Kupfer(II)-komplex-Gemisch ein IR-Spektrum auf, das dem folgenden entsprechen sollte. Vergleichen Sie es mit dem IR-Spektrum des N-meso-Diastereoisomer (unten).



Nehmen Sie vom Nickel(II)-komplex-Gemisch ein Festkörper-UV-Spektrum auf, das dem folgenden entsprechen sollte. Interpretieren Sie es.
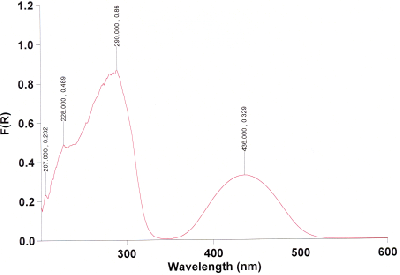
Zum Vergleich: Das UV-Spektrum des gemischten Komplexes aus wässriger Lösung.
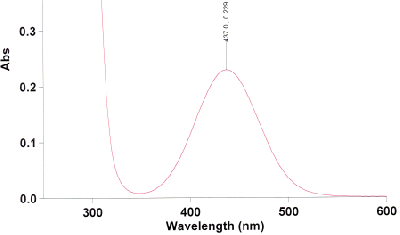
Literatur: Synthese von Antimon(III)-iodid: Brauer, 3. Auflage, Band I, S. 591. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek Chemie: VH 5501 B825; zum chemischen Transport von Sb2S3: H. Schäfer, H. Plautz, C. Balarew, J. Bazelkov, Z. Anorg. Allg. Chem. 1978, 440, 130--136; B. Vengatesan, N. Kanniah, P. Ramasamy, Mater. Chem. Phys. 1987, 17, 311--316; C. Balarew, M. Ivanova, Cryst. Res. Technol. 1986, 21, K171--K175; J. Yang, Y.-C. Liu, H.-M. Lin, C.-C. Chen, Adv. Mater. 2004, 16, 713--716.
Benötigte Chemikalien: Iod, Toluol, CaCl2 als Trockenmittel, Antimon (Pulver), Schwefel.
Darstellung von Antimon(III)-iodid, SbI3
3,5 g Iod (14 mmol) werden in 75 mL Toluol (Kp 110 °C) gelöst (Toluol muss zuvor wenigstens 24 Stunden über CaCl2 getrocknet werden) und mit 1,8 g feingepulvertem Antimon (15 mmol) unter Rückfluss bei ca. 120 °C erhitzt bis zum Verschwinden der Iodfarbe im kondensierten Toluol. Die grünlichgelbe Lösung wird von nicht umgesetzten Antimon heiß unter N2-Atmosphäre in einer Schlenkfritte abgetrennt (Assistent!). Dabei muss die Schlenkfritte sehr gut vorgeheizt werden mit einer Heißluftpistole, da ansonsten viel Produkt in der Fritte hängen bleibt. Das Produkt kristallisiert beim Abkühlen sehr schnell im Filtratkolben aus. Zur Erhöhung der Ausbeute wird der abgekühlte Filtratkolben über Nacht im Kühlschrank aufbewahrt. Anschließend wird unter N2-Atmosphäre abgefrittet und das Produkt in einen kleinen Rundkolben überführt. Das Produkt wird im Vakuum getrocknet (kann bereits mit dem noch in der Fritte befindlichen Produkt geschehen, Kühlfalle!).
Anmerkung für die Assistenten: Die sich als rein erweisenden SbI3-Fraktionen aller Versuchsgruppen werden in einem einzigen Kolben gesammelt. Die verunreinigten Fraktionen werden sachgerecht entsorgt.
Darstellung von Antimon(III)-sulfid, Sb2S3
Wiegen Sie Antimon und Schwefel in stöchiometrischer Menge und einer Gesamtmasse von 0,20 g sowie 0,02 g Antimon(III)-iodid ab
und zermörsern Sie alles zu einem sehr feinen, homogenen Gemisch. Dann bringen Sie das Gemisch mit Hilfe eines langen Trichters
an das geschlossene Ende einer zuvor unter Vakuum ausgeheizten Duranglasampulle.
Anschließend wird die Ampulle evakuiert und unter Anleitung eines Assistenten mit einem Gebläsebrenner abgeschmolzen. Achten Sie
darauf, dass die Substanzmischung immer am gleichen Ende der Ampulle bleibt und es nicht zu Verunreinigungen im restlichen Teil
der Ampulle kommt.
Bringen Sie die Ampulle geschützt durch ein Korundrohr in einen geeigneten Ofen, so dass sich das Substanzgemisch bei einer
Temperatur von 490 °C befindet und das andere Ende der Ampulle bei etwa 410 °C (die Temperatur darf auch unter 410 °C).
Hierzu muss das Temperaturprofil
des Ofens bekannt sein. Beginnen Sie rechtzeitig mit dessen Bestimmung.
Lassen Sie die Ampulle wenigstens 24 Stunden im Ofen.
In einem Testversuch wurden in einer etwa 16 cm langen Ampulle nach zwei Tagen sehr schöne Kristalle erhalten, wobei etwa die
Hälfte der ursprünglich eingesetzten Substanzmenge transportiert wurde.
Wenn Sie die Reaktion mit dem Carbolite 12/65/550 Rohrofen durchführen, beachten Sie bitte folgende Hinweise: Die Zieltemperatur
sollte 505 °C sein. In der Regel wird der Ofen bereits in Betrieb sein und auf diese Temperatur eingestellt sein. Den
betreitliegenden Temperaturfühler können Sie zur Temperaturmessung verwenden. Achten Sie darauf, dass Sie die Temperatur
innerhalb des Schutzrohres messen. Während Sie das Ofenprofil aufnehmen, muss der Ofen mit Ofenwolle verschlossen sein!
Schieben Sie die Glaswolle nicht zu weit in das Rohr hinein (maximal 1--2 cm). Wenn
Sie das Temperaturprofil so gut kennen, dass Sie wissen, wie weit Sie die Ampulle in den Ofen schieben müssen, damit das
Substanzgemisch bei etwa 490 °C liegt und ein anderer Bereich der Ampulle gleichzeitig bei etwa 410 °C, dann nehmen Sie
den Temperaturfühler aus dem Ofen und schalten ihn aus und bringen dann die Ampulle in die richtige Position. Dazu gehen Sie am
besten wie folgt vor: Messen Sie den Abstand vom Substanzgemisch zum späteren kalten Ende der Ampulle. Ziehen Sie dann das
Korundrohr etwas aus dem Ofen raus, schieben Sie die Ampulle so in das Korundrohr, dass das spätere kalte Ende bündig zum
Korundrohrende liegt, und schieben Sie dann das Korundrohr so weit wie eben nötig in den Ofen hinein. Schließen Sie dann den
Ofen mit Ofenwolle. Achten Sie dabei darauf, dass Sie das Korundrohr nicht verschieben. Die Ofenwolle muss nicht weit in das
Rohr gesteckt werden, allerdings so weit, wie auch bei der Bestimmung des Ofenprofils.
Lassen Sie von Antimon(III)-iodid ein Massenspektrum aufnehmen und interpretieren Sie das Spektrum.
Das erhaltene Massenspektrum sollte dem folgenden entsprechen:
Das erhaltene Antimon(III)-sulfid könnte mit Einkristall- oder Pulverdiffraktometrie oder per ICP untersucht werden. Bitte mit Assistenten abklären.
Gehen Sie im Protokoll auf die Temperaturabhängigkeit der Transportrichtung ein (Vergleich Sb2S3 mit Mondreaktion Ni(Ni(CO)4 oder mit einer Sublimation).
Literatur: zur Synthese: Jander-Blasius, Lehrbuch der analytischen und präparativen anorganischen Chemie, 16. Aufl., Hirzel Verlag, Stuttgart, 2006, Seite 208--209. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek Chemie: VH 5100 J33; allgemeine Übersicht in Chemie in unserer Zeit 2002, 36, 108-124.
Vorsicht: Das Zermörsern des Substanzgemischs unter dem Abzug durchführen!
Benötigte Chemikalien: Yttrium(III)-oxid, Bariumperoxid, Kupfer(II)-oxid.
In einem trockenen Achatmörser werden 0,2 g Yttrium(III)-oxid und die nötigen stöchiometrischen Mengen Bariumperoxid und
Kupfer(II)-oxid gründlich gemörsert (10 Minuten!), so dass am Ende eine homogene Mischung vorliegt, in der keine sichtbaren
Körnchen einer Einzelkomponente mehr zu erkennen sind.
Aus dem Gemisch wird eine Menge von 0,5 g Material bei einem Druck von 10 t in Tablettenform gepresst. Die Tabletten müssen
vorsichtig gehandhabt werden, da sie leicht brechen. Wenn sie brechen, muss die Substanz erneut gemörsert werden.
Die Tabletten werden vorsichtig auf eine Korundplatte gelegt und im Ofen innerhalb von 16 Stunden auf 930 °C erhitzt.
Danach wird die Probe weitere 16 Stunden bei dieser Temperatur gehalten. Dann wird innerhalb von 5 Stunden auf 450 °C
abgekühlt und die Probe wiederum 16 Stunden bei dieser Temperatur gehalten. Danach sollten die Tabletten ein dunkelgrau-schwarzes
Aussehen besitzen.
Nachweis des Meissner-Effekts
Die Tablette und eine Ringmagnet werden in flüssigem Stickstoff vorsichtig abgekühlt. Anschließend legt man den Ringmagnet
mit einer Plastikpinzette auf eine Styroporunterlage und die Supraleitertablette vorsichtig darüber. Solange die
Tablette und der Magnet ausreichend kalt sind (schnell arbeiten!), sollte die Tablette frei über dem
Magneten schweben.
Erläutern Sie das Phänomen der Supraleitung und den hoffentlich beobachteten Meissner-Effekt. Beschreiben Sie auch die Struktur dieser Supraleiter.
Literatur: allgemein z.B.: M. Hansen, Constitution of binary alloys, McGraw Hill Co., New York, 1958; Phasendiagramme: W. O. Moffatt, Handbook of binary phase diagrams, General Electric Corp. 1981; Hanson, Binary phase diagrams.
Benötigte Chemikalien: Kupfer, Zink, Kaliumchlorid, Natriumchlorid, wasserfreies Calciumchlorid, Cadmium.
Allgemeine Darstellungsvorschrift
Die Synthese intermetallischer lässt sich im Labor am einfachsten durch Zusammenschmelzen der Elemente im stöchiometrischen
Verhältnis erreichen. Die Substanzmengen werden so gewählt, dass die Ansatzgröße zwischen 10 g und 15 g liegt. Wird eine Schutzschmelze
zur Vermeidung von Oxidation oder Verdampfung benötigt, so wird etwa 15 g bis 20 g Salzmischung hierfür verwendet.
Nach dem Erkalten der Schmelze können die Salze der Schutzschmelze mit heißem Wasser gelöst werden. Verunreinigungen werden
abgefeilt. Von der gewünschten Probe wird etwas Substanz abgefeilt für ein Pulverdiffraktogramm.
Cu2Zn, α-Messing
Für die Schutzschmelze werden 20 g Kaliumchlorid verwendet. Das Gemisch wird bei 1000 °C 2 Stunden geschmolzen, dann bei
850 °C 2 Tage lang getempert. Danach wird schnell abgekühlt.
Cu5Zn8, γ-Messing
Für die Schutzschmelze wird eine 1:2-Mischung aus NaCl und CaCl2 verwendet.
Das Gemisch wird bei 950 °C 2 Stunden geschmolzen, dann nach Abschrecken auf Raumtemperatur bei
750 °C 12 Stunden getempert.
Cu3Sn, ε-Bronze
Für die Schutzschmelze wird eine 1:2-Mischung aus NaCl und CaCl2 verwendet.
Das Gemisch wird bei 850 °C 2 Stunden geschmolzen, dann nach Abschrecken auf Raumtemperatur bei
640 °C 12 Stunden getempert.
Cu4Cd3
Für die Schutzschmelze wird eine 3:1-Mischung aus KCl und CaCl2 verwendet.
Das Gemisch wird bei 700 °C 1 Stunden geschmolzen, dann bei
450--500 °C 3 Stunden getempert.
Literatur: allgemein: Gmelin, Jander-Blasius, Brauer.
Benötigte Chemikalien: Zinn(IV)-oxid, Bismut(III)-oxid, Blei(II)-oxid, Blei(II)-sulfid, Natriumcarbonat, Kaliumcarbonat, Kaliumcyanid.
Vorsicht: Kaliumcyanid darf nicht im Saal gelagert werden. Erforderliche Substanzmenge bei Frau Peter holen. Entsorgung cyanidhaltiger Abfälle in gesondertem Behälter!
Reduktion von Metalloxiden mit Kaliumcyanid
Das Metalloxid wird in einem unglasierten Porzellantiegel mit einem Überschuss Kaliumcyanid (7 g Zinn(IV)-oxid/5 g Kaliumcyanid,
3 g Blei(II)-oxid/3 g Kaliumcyanid, 10 g Bismut(III)-oxid/5 g Kaliumcyanid) 5 Minuten mit einem Gebläsebrenner erhitzt (30 Minuten
mit dem Bunsenbrenner). Nach dem Erkalten wird der Schmelzkuchen mit Wasser ausgelaugt.
Röstreaktionsarbeit zur Darstellung von Blei
12 g Blei(II)-sulfid und 22 g Blei(II)-oxid werden gut vermischt in einem unglasierten Porzellantiegel unter einer Schutzschicht
aus Natriumcarbonat/Kaliumcarbonat mit einem Gebläsebrenner ca. 30 Minuten lang auf dunkle Rotglut erhitzt. Anschließend wird noch
10 Minuten lang auf helle Rotglut erhitzt. Dabei sollte öfters mit einem Magnesiastäbchen umgerührt werden. Nach dem
Erkalten wird der Schmelzkuchen mit Wasser ausgelaugt.
Charakterisieren Sie die Elemente mit einem Pulverdiffraktogramm.
Literatur: SrTiO3: Z. Phys. Chem. 1935, B28, 65; Gmelin, Standardlehrbücher, Sol-Gel-Verfahren: M. Kakihana, T. Okubo, M. Arima, Journal of Sol-Gel Science and Technology 1998, 12 95.
Benötigte Chemikalien: Strontiumcarbonat, Titandioxid, Cobalt(II)-carbonat, Aluminium(III)-oxid, Kaliumchlorid, Silbernitratlösung, Aerosil (Siliziumdioxid), Bismut(III)-oxid, Strontiumnitrat, Methanol, Tetraethoxysilan, .
Darstellung des Perowskits SrTiO3
4,02 g Strontiumcarbonat und 2,18 g Titandioxid werden innig miteinander verrieben und in einem Porzellantiegel zunächst
24 Stunden bei 650 °C geglüht, dann 24 Stunden bei 800 °C und schließlich drei Tage bei 1000 °C. Zwischen den
einzelnen Schritten wird das Gemenge immer wieder ordentlich gemörsert.
Darstellung des Spinells CoAl2O4
3,4 g Cobalt(II)-carbonat und 2,9 g Aluminium(III)-oxid werden mit 9,4 g Kaliumchlorid vermischt und in einem Porzellantiegel
im Ofen zunächst zwei Stunden auf 500 °C erhitzt (Deckel mit Loch verwenden!). Anschließend wird etwa 15 Stunden auf
1100 °C erhitzt. Danach wird durch mehrfaches Auslaugen mit kochendem Wasser das Kaliumchlorid entfernt. Das Filtrat wird
mit Silbernitratlösung auf Vohandensein von Chlorid geprüft.
Darstellung der Silikate Bi4(SiO4)3 und SrSiO3
a) Bi4(SiO4)3: Zunächst werden einige Gramm Aerosil in einen unglasierten Porzellantiegel
gegeben und bis zur Gewichtskonstanz bei 450 °C im Ofen entwässert. Einige Gramm Bismut(III)-oxid werden über Nacht
im Trockenschrank getrocknet. Dann werden stöchiometrische Mengen an Aerosil und Bismut(III)-oxid (Gesamtmenge etwa 3 g) in einer
Reibschale verrieben und im Tiegel eine Stunde auf 800 °C erhitzt. Danach lässt man abkühlen, zermörsert gründlich und
stellt den Tiegel zwei Stunden bei 800 °C in den Ofen. Danach lässt man die Probe wieder abkühlen, zermörsert gründlich
und erhitzt nochmals 3 Stunden auf 800 °C.
b) SrSiO3 nach dem Sol-Gel-Verfahren: Es werden 6,47 g Strontiumnitrat in soviel Wasser gelöst, dass alles gut
gelöst ist, aber keine gesättigte Lösung vorliegt. Anschließend wird soviel Methanol zugegeben, dass eine Mischung mit einem
Wasser/Methanol-Verhältnis von ca. 1:2 entsteht. Dabei darf das Salz nicht ausfallen (notfalls etwas Wasser zugeben) und die
Lösungsmittelmenge sollte nicht zu groß sein, da das Lösungsmittel restlos eingedampft werden muss. Zu dieser Lösung werden
6,84 mL Tetraethoxysilan gegeben, wobei sich keine Öltröpfchen auf der Oberfläche bilden dürfen (notfalls Methanol zusetzen).
Zur vollständigen Hydrolyse wird das Gefäß mit einem Uhrglas und Parafilm verschlossen mindestens 24 Stunden lang bei Raumtemperatur stehen
gelassen (darf auch länger sein). Danach wird die Mischung auf dem Wasserbad unter gelegentlichem Rühren bis zur Trockne
eingedampft. Wichtig: das Tetraethoxysilan muss vor dem Eindampfen vollständig hydrolysiert sein, da es ansonsten abdampft!
Das entstandene Pulver wird im Trockenschrank über Nacht bei 110--130 °C getrocknet und anschließend zuerst eine Stunde lang bei
300--400 °C geglüht, dann nochmals vier Stunden lang bei 1000 °C. Man erhält ein weißes Pulver.
Charakterisieren Sie die Verbindungen mit einem Pulverdiffraktogramm.
Literatur: Inorganic Synthesis 25, 135--139. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek Chemie: VH 5500.
Benötigte Chemikalien: Kaliumhydrogencarbonat, Wasser, Cobalt(II)-chlorid, Wasserstoffperoxid (30%ig), Glycin, Eisessig, Ethanol (zum Waschen), Diethylether (zum Waschen).
9,00 g Kaliumhydrogencarbonat (90 mmol) werden in einem 50 mL Erlenmeyerkolben zu 9 mL Wasser
gegeben und in einem Eisbad 15 Minuten lang gerührt (Mischung A, man erhält keine Lösung!).
In einem 50 mL Rundkolben werden 3,06 g Cobalt(II)-chlorid (12,6 mmol) in 3 mL Wasser gelöst
bei maximal 30 °C. Diese Mischung wird dann 15 Minuten lang in ein Eisbad gestellt, worauf
dann 4,5 mL 30%iges Wasserstoffperoxid langsam tropfenweise zugegeben wird (Lösung B).
Dann wird Lösung B tropfenweise (alle 5 sec ein Tropfen) unter Eiskühlung und rühren zur Mischung A
gegeben. Danach wird die Mischung filtriert und gibt zum Filtrat in einem 100 mL Erlenmeyerkolben
2,70 g Glycin (36 mmol), erhitzt die resultierende Lösung unter Rühren etwa 30 Minuten lang im Ölbad
auf 60--70 °C, bis die Lösung eine dunkelblaue bis violette Farbe angenommen hat.
Daraufhin gibt man bei 60--70 °C unter heftigem Rühren tropfenweise (alle 5 sec ein Tropfen) 6,3 mL
6 molare Essigsäure hinzu. Zur Vervollständigung der Reaktion können noch 0,15 mL 6 molare Essigsäure
zugefügt werden, ehe man heftig rührt bis die Kohlendioxidbildung aufhört und die Lösung eine rötlich
violette Farbe angenommen hat. Diese Lösung wird am Rotavapor so lange eingeengt bis der erste Niederschlag
auftritt. Dieser wird durch Zugabe von Wasser gerade wieder aufgelöst und die gesamte Lösung wird über
Nacht bei Raumtemperatur stehen gelassen. Der dann ausgefallene pink-rot-farbene Niederschlag (fac-Isomer) wird in
einer Glasfritte (Porengröße 3) abfiltriert und nacheinander mit je 3 mL Wasser, Ethanol und Diethylether gewaschen
und anschließend 2 Stunden bei 100 °C im Vakuum getrocknet. Das Filtrat wird am Rotavapor eingeengt bis die ersten
violetten Kristalle vom besser löslichen mer-Isomer ausfallen. Dann lässt man den Kolben über Nacht stehen
und filtriert am nächsten Tag in einer Glasfritte (Porengröße 3) ab und wäscht den Niederschlag nacheinander
mit je 3 mL Wasser, Ethanol und Diethylether, ehe er 2 Stunden bei 100 °C im Vakuum getrocknet wird.
Im Laufe der Synthese treten zahlreiche Farbänderungen auf. Ordnen Sie im Protokoll den verschiedenen (Zwischen)-Produkten die richtige Farbe zu und begründen Sie die Farben und Farbänderungen.
Versuchen Sie auf Papier und mit Hilfe des bereit gestellten Molekülbaukastens abzuleiten, wie viele verschiedene Isomere es von Tris(glycinato)cobalt(III) geben kann. Beachten Sie dabei, dass ein Glycin-Molekül niemals zwei trans-ständige Koordinationsstellen am Cobalt(III)-Zentrum besetzen kann.
Nehmen Sie von den beiden Produkten UV/Vis-Spektren auf und interpretieren Sie diese (10 mL einer 0,5 mM Lösung des fac-Isomers und 10 mL einer 5 mM Lösung des mer-Isomers herstellen und messen). Vergleichen Sie auch die experimentell bestimmten Maxima mit den in der Literatur angegebenen.
Die erhaltenen UV/Vis-Spektren sollten demn folgenden entsprechen (oben fac-Isomer, unten mer-Isomer):
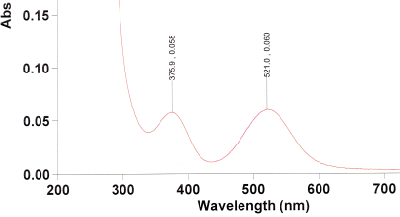
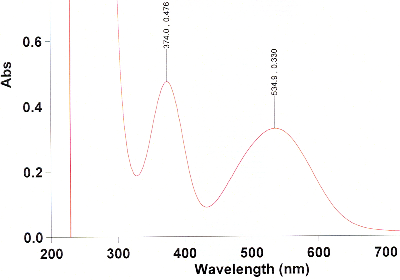
Literatur: Jander-Blasius, Lehrbuch der analytischen und präparativen anorganischen Chemie, 16. Aufl., Hirzel Verlag, Stuttgart, 2006, Seite 201--202. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek Chemie: VH 5100 J33
Vorsicht: Kaliumdichromat ist giftig! Schutzhandschuhe tragen!
Benötigte Chemikalien: Aluminiumsulfat 18-Hydrat, Kaliumsulfat, Kaliumdichromat, konz. Schwefelsäure, Ethanol.
Synthese von Aluminiumalaun
9,9 g Aluminiumsulfat 18-Hydrat werden in 7,5 mL heißem Wasser gelöst und eine warme Lösung
von 2,55 g Kaliumsulfat in 15 mL Wasser zugefügt. Die Lösung lässt man möglichst langsam
und erschütterungsfrei abkühlen, wobei sich der Alaun in farblosen Kristallen abscheidet. Nach zwei, drei Tagen
filtriert man die Kristalle ab und lässt sie trocknen.
Synthese von Chromalaun
Eine Lösung von 5,0 g Kaliumdichromat in 50 mL Wasser und 5,5 mL konz. Schwefelsäure wird tropfenweise mit
3,5 mL Ethanol versetzt (eventuell kühlen mit Eisbad, Thermometer, Temperatur sollte um die 30 °C liegen und
darf nicht über 40 °C steigen). Beim erschütterungsfreien offenen Stehenlassen
kristallisiert der Chromalaun. Nach zwei, drei Tagen filtriert man die Kristalle ab und trocknet sie auf Wischpapier durch
gründliches Abtupfen. Dann löst man die Kristalle unter leichtem Erwärmen (max. 40 °C) in wenig Wasser (für 3 g Substanz
etwa 12 mL Wasser) und lässt langsam abkühlen. Tags darauf solllten sich Kristalle mit gut ausgebildeten Kristallflächen
gebildet haben. Diese lässt man auf Wischpapier gut trocknen.
Geben Sie Ihre Kristalle von Aluminiumalaun und Chromalaun dem/der zuständigen Praktikumsassistenten/in. Diese(r) wird Ihnen dann
eine Mischung der beiden Alaune aushändigen, deren Zusammensetzung Sie per UV-Spektroskopie und per Pulverdiffraktometrie
bestimmen sollen.
Mischkristalle
2,00 g der Mischung mit unbekannter Zusammensetzung werden unter Erwärmen in einem kleinen Becherglas in 9 mL Wasser gelöst
und dann langsam abgekühlt auf Raumtemperatur. Zur Beschleunigung der Kristallisation kann man den Boden des Becherglases
mit einem Glasstab ankratzen. Die Kristalle werden abfiltriert und dann nochmals aus Wasser umkristallisiert. Danach werden
die Kristalle an Luft getrocknet. Dann wird zermörsert und ein
Pulverdiffraktogramm in Auftrag gegeben.
Nachfolgend sehen Sie die Pulverdiffraktogramme und die zugehörige Liste mit d-Werten von Aluminiumalaun (oben) und Chromalaun (unten). Indizieren Sie die Diffraktogramme und berechnen Sie die Gitterkonstanten der beiden kubisch kristallisierenden Alaune. Das Diffraktogramm wurde mit Cu-Kα1-Strahlung gemessen (Wellenlänge λ = 1,5405 Angstrom).
Aluminiumalaun:

Chromalaun:

Lassen Sie von der Ihnen ausgehändigten Mischung aus Aluminium- und Chromalaun ein Pulverdiffraktogramm aufnehmen und indizieren Sie das Diagramm. Berechnen Sie die Gitterkonstante und geben Sie den Gehalt an Chromalaun in der Mischung an. Gehen Sie dabei davon aus, dass die Vegard'sche Regel gilt.
Den oben röntgenographisch bestimmten Gehalt an Chromalaun in der Ihnen ausgehändigten Mischung versuchen Sie
jetzt UV-spektroskopisch zu bestätigen. Dazu gehen Sie wie folgt vor:
Zunächst stellen Sie drei wässrige Lösungen von je 10 mL her, die jeweils eine Einwaage von 0,200 g enthalten, und zwar
a) 0,200 g Chromalaun, 0 g Aluminiumalaun, b) 0,140 g Chromalaun, 0,060 g Aluminiumalaun und
c) 0,060 g Chromalaun, 0,140 g Aluminiumalaun. Die drei Mischungen werden jeweils in 10 mL Messkolben auf 10 mL aufgefüllt.
Diese drei Lösungen dienen zur Erstellung einer Eichgeraden. Mit Hilfe der Eichgeraden bestimmen Sie den Gehalt an
Chromalaun in der Ihnen ausgehändigten Mischung. Wiegen Sie von dieser Mischung 0,200 g in einen 10 mL Messkolben
und füllen Sie auf 10 mL mit Wasser auf.
Zuerst messen Sie die Baseline von reinem Wasser. Dann nehmen Sie das UV-Spektrum von reinem Chromalaun auf. Verwenden Sie dazu
nicht Lösung a) (die brauchen Sie später noch), sondern lösen Sie einfach etwas Chromalaun in Wasser, so dass eine nicht allzu
stark gefärbte Lösung entsteht. Drucken Sie das erhaltene Spektrum in eine pdf-Datei und fügen Sie es dem Protokoll bei.
Diese Messung dient der Bestimmung von Absorptionsmaxima zur Konzentrationsmessung. Es gibt im Spektrum (siehe unten) zwei peaks,
die sich dazu eignen (408 nm und 575 nm). Sie entscheiden sich für 408 nm. Warum?
Schließen Sie das Programm, starten Sie es neu, laden Sie die zuvor gemessene Baseline und messen Sie das Spektrum von
Aluminumalaun, indem Sie einfach etwas Aluminiumalaun in Wasser lösen und in die Küvette bringen. Sie sollten ein Spektrum erhalten,
das dem unten abgebildeten ähnelt. Drucken Sie ihr Spektrum in eine pdf-Datei und fügen Sie es dem Protokoll bei.
Schließen Sie das Programm.
Beurteilen Sie, in welchem Ausmaß Aluminiumalaun das Spektrum von Chromalaun verfälscht?
Starten Sie das Programm CaryWinUV durch Doppelklick auf das entsprechende Icon. Dann Doppelklick auf das Icon Concentration.
Im sich öffnenden Programmfenster drücken Sie Setup und tragen dann unter Cary die Wellenlänge 408 nm ein (oder den
unter Umständen leicht davon abweichenden Wert, den Sie zuvor bestimmt haben). Unter Standards setzen Sie Unit auf mg/mL und
setzen die Zahl der Standards auf 3. Dann editieren Sie für Standard 1 Concentration 6, für Standard 2 Concentration 14 und für
Standard 3 Concentration 20. Sie messen also nachher von der verdünnten zur konzentrierteren Chromalaunlösung. Den Fit type belassen
Sie auf linear. Dann setzen Sie die Zahl der Samples auf 1 (das wird ihre Probe mit unbekannter Zusammensetzung sein).
Unter Report tragen Sie unter Name ihre Namen ein und unter Comment halten Sie nochmals die Zusammensetzung der Standards fest.
Dann gehen Sie mit ok raus.
Dann wird zunächst die Absorption des reinen Lösungsmittel Wasser gemessen. Füllen Sie die Küvette mit Wasser und setzen Sie sie
ein. Drücken Sie den Zero Button, dann ok. Wenn die Messung abgeschlossen ist, steht oben links unter Abs 0.001 und oben rechts
unter Nm 408 (oder der Wert, den Sie eingegeben haben).
Nehmen Sie die Küvette raus und spülen Sie sie mit etwas Lösung von Standard 1. Dann füllen Sie die Küvette mit der Lösung von
Standard 1 und setzen Sie ein. Drücken Sie Start, klicken Sie Standard 1 an und drücken ok. Speichern Sie die Daten unter
Austausch\LAF_UV\Cary50 unter dem Namen chromalaun_standard1_gn.bcn (gn steht für ihre Gruppennummer). Drücken Sie save,
dann ok, worauf Standard 1 gemessen wird. Beachten Sie die Angaben zur Messung im Reportfenster.
Nehmen Sie die Küvette raus und spülen Sie sie mit etwas Lösung von Standard 2. Dann füllen Sie die Küvette mit der Lösung von
Standard 2 und setzen Sie ein. Drücken Sie ok. Standard 2 wird gemessen. Beachten Sie die Angaben zur Messung im
Reportfenster.
Nehmen Sie die Küvette raus und spülen Sie sie mit etwas Lösung von Standard 3. Dann füllen Sie die Küvette mit der Lösung von
Standard 3 und setzen Sie ein. Drücken Sie ok. Standard 3 wird gemessen. Beachten Sie die Angaben zur Messung im
Reportfenster.
Dann kommt die Aufforderung zur Messung von Sample 1. Nehmen Sie die Küvette raus und spülen Sie sie mit etwas Lösung von
ihrer zu untersuchenden Substanz. Dann füllen Sie die Küvette mit der Lösung von
ihrer zu untersuchenden Substanz und setzen Sie ein. Drücken Sie ok. Sample 1 wird gemessen. Drucken Sie das Ergebnis dann
in die Datei chromalaun_uv_conc_gn.pdf unter Austausch\LAF_UV\Cary50. Im Reportfenster wird die Konzentration an Chromalaun
in ihrer zu untersuchenden Mischung angegeben. Sie sollte mit der röntgenographisch ermittelten übereinstimmen. In einem
Testversuch haben wir 110 mg Chromalaun und 90 mg Aluminiumalaun eingewogen. Das Ergebnis (Eichgerade und Gehalt der Mischung
sehen Sie unten abgebildet.
Warum geht die Eichgerade nicht durch den Ursprung?
Das UV-Spektrum von Chromalaun in wässriger Lösung:
2_uv.png)
Das UV-Spektrum von Aluminiumalaun in wässriger Lösung:
2_uv.png)
Die Eichgerade im Testversuch:
Die Angaben zur Messung der Standards und der zu untersuchenden Mischung im Testversuch:


Literatur: Inorganic Synthesis, 24, 183--184. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek Chemie: VH 5500.
Vorsicht: Im Nachtlabor aufbauen!
Benötigte Chemikalien: Chrom(III)-chlorid Hexahydrat, Wasser, Ethanol, Harnstoff, 2,2,6,6-Tetramethyl-3,5-heptanedion
0,75 g Chrom(III)-chlorid Hexahydrat werden in einer Mischung aus 6,3 mL Wasser und 16,3 mL
Ethanol gelöst. Zu dieser Lösung werden 5,0 g Harnstoff und 1,25 g 2,2,6,6-Tetramethyl-3,5-heptanedion
gegeben. Diese Mischung wird unter Rühren 24 h unter Rückfluss zum Sieden erhitzt. Die Farbe der
Lösung sollte sich in dieser Zeit von tiefgrün zu purpur ändern und ein dunkler Niederschlag sollte ausfallen.
Man lässt die Lösung auf Raumtemperatur abkühlen und verdünnt dann durch Zugabe von 25 mL Wasser.
Das feste Produkt wird abfiltriert und dreimal mit je 12,5 mL Wasser gewaschen. Der Niederschlag wird
an Luft bei Raumtemperatur getrocknet und dann zur Reinigung bei 180 °C und 0,1 mbar Druck sublimiert.
Dabei wird eine spezielle Sublimationsapparatur verwendet, die zunächst zusammen mit dem Rohprodukt evakuiert wird
auf einen Druck von etwas unter 0,1 mbar. Dann schließt man den Hahn zur HV und taucht den Kolben in ein auf 180 °C
vorgeheiztes Ölbad. Hin und wieder wird durch öffnen des Hahns zur HV der Druck überprüft. Das gewünschte Produkt
ist braunviolett. Wenn die Reaktionsbedingungen eingehalten werden, sublimiert kein hellgrünes Produkt über.
Das Produkt ist nicht luftempfindlich.
Anmerkung für die Assistenten: Die sich als rein erweisenden Fraktionen aller Versuchsgruppen werden in einem einzigen Kolben gesammelt. Die verunreinigten Fraktionen werden sachgerecht entsorgt.
Lassen Sie ein IR-Spektrum der Verbindung aufnehmen und interpretieren Sie dieses (es sollte dem nachfolgend abgebildeten ähneln). Vergleichen Sie es mit dem des Tris(acetylacetonato)chrom(III)-Komplexes.
Das IR-Spektrum von Tris(2,2,6,6-tetramethyl-3,5-heptanedionato)chrom(III):
_ir_spektrum.png)
Peakangaben zum IR-Spektrum von Tris(2,2,6,6-tetramethyl-3,5-heptanedionato)chrom(III):
_ir_peaks.png)
Literatur: C. Glidewell in Inorganic Experiments, Wiley VCH, Weinheim, 2003, Seite 149--159. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek Chemie: VH 5100 W913(2).
Benötigte Chemikalien: Acetylaceton, Ammoniumhydroxid, Aluminiumsulfat 18-Hydrat, Wasser.
In einem 100 mL Erlenmeyerkolben werden 3,1 mL Acetylaceton versetzt mit 40 mL Wasser und 8 mL 5 molare Ammoniumhydroxidlösung.
3,33 g Aluminiumsulfat 18-Hydrat werden in 30 mL kaltem Wasser gelöst. Zu dieser Lösung wird die ammoniakalische Acetylacetonlösung
portionsweise und unter Schütteln gegeben. Danach prüft man den pH-Wert und gibt, falls die Lösung noch sauer ist, kleine Portionen
von 5 molarer Ammoniumhydroxidlösung hinzu, bis die Lösung pH-neutral ist. Dann lässt man noch 15 Minuten stehen und filtriert dann
den cremefarbenen Niederschlag ab und wäscht diesen mit 100 mL kaltem Wasser und saugt den Niederschlag 10 Minuten lang trocken an
der Wasserstrahlpumpe. Danach wird der Niederschlag im Vakuum getrocknet.
Lassen Sie ein 1H- und ein 13C-NMR-Spektrum aufnehmen von dem Aluminiumkomplex (Lösungsmittel CDCl3). Interpretieren Sie die Spektren und vergleichen Sie sie mit den entsprechenden Spektren von reinem Acetylaceton in CDCl3.
Das 13C-NMR-Spektrum von Tris(acetylacetonato)aluminium(III) in CDCl3:
3_13c_nmr.png)
Das 13C-NMR-Spektrum von Acetylaceton in CDCl3:
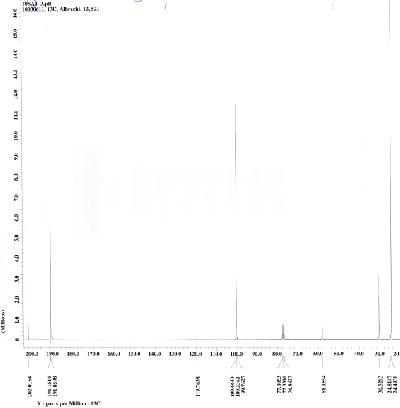
Das 1H-NMR-Spektrum von Tris(acetylacetonato)aluminium(III) in CDCl3:
3_1h_nmr.png)
Das 1H-NMR-Spektrum von Acetylaceton in CDCl3:

Nehmen Sie ein IR-Spektrum des Komplexes auf und interpretieren Sie es. Vergleichen Sie es mit dem IR-Spektrum von reinem Acetylaceton (unten).
Das IR-Spektrum von Tris(acetylacetonato)aluminium(III):
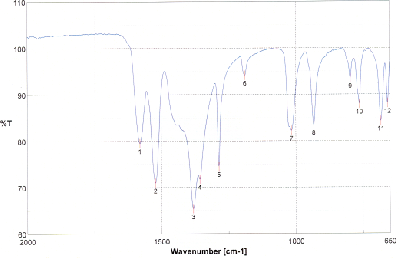
Peakangaben zum IR-Spektrum von Tris(acetylacetonato)aluminium(III):

Das IR-Spektrum von Acetylaceton:
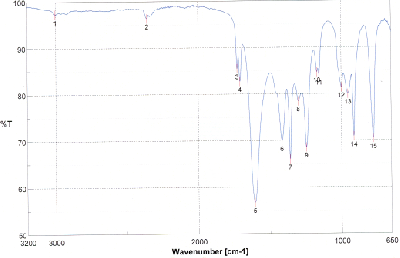
Peakangaben zum IR-Spektrum von Acetylaceton:

Literatur: C. Glidewell in Inorganic Experiments, Wiley VCH, Weinheim, 2003, Seite 149--159. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek Chemie: VH 5100 W913(2).
Benötigte Chemikalien: Chrom(III)-chlorid Hexahydrat, Acetylaceton, Harnstoff, Wasser.
In einem 100 mL Erlenmeyerkolben werden 1,4 g Chrom(III)-chlorid Hexahydrat in 50 mL Wasser gelöst. Dazu gibt man 10 g Harnstoff
in drei oder vier Portionen, wobei man nach jeder Zugabe gut schüttelt. Dann gibt man zu dieser Mischung 3,1 mL Acetylaceton
mit einer Pipette tropfenweise zu. Die Mischung wird gut geschüttelt, mit einem Uhrglas bedeckt und in einem auf 90 °C
vorgeheizten (!!) Ölbad schnell auf 80--90 °C aufgeheizt. Kontrollieren Sie die Temperatur in der Lösung immer wieder mit einem
Thermometer. Nach 90 Minuten bei 80--90 °C entfernt man das Ölbad und lässt die Mischung, in der sich jetzt dunkelbraunviolette
Kristalle befinden, auf Raumtemperatur abkühlen. Es wird abfiltriert und die Kristalle ungewaschen an Luft getrocknet.
Nehmen Sie ein IR-Spektrum des Komplexes auf und interpretieren Sie es. Vergleichen Sie es mit dem von Tris(acetylacetonato)aluminium(III).
Das IR-Spektrum von Tris(acetylacetonato)chrom(III):
3_ir_spektrum.png)
Peakangaben zum IR-Spektrum von Tris(acetylacetonato)chrom(III):
3_ir_peaks.png)
Warum können Sie von Tris(acetylacetonato)chrom(III) keine NMR-Spektren aufnehmen (sie könnten schon, aber warum macht es keinen Sinn?), wohl aber von Tris(acetylacetonato)aluminium(III)?
Das UV-Spektrum von Tris(acetylacetonato)chrom(III) gelöst in Ethanol (die Substanz ist in Wasser kaum löslich):
3_uv_etoh.png)
Literatur: Inorg. Synth. 25, 127--129. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek Chemie: VH 5500.
Vorsicht: Versuch unbedingt im Abzug durchführen!
Benötigte Chemikalien: Eisen(II)-sulfat Heptahydrat, Natriumnitrit, Natriumdiethyldithiocarbamat, verdünnte Schwefelsäure, Chloroform, wasserfreies Magnesiumsulfat, Petrolether.
2,22 g Eisen(II)-sulfat Heptahydrat und 0,38 g Natriumnitrit werden in 7,5 mL verdünnter Schwefelsäure
gelöst und dann 2,50 g Natriumdiethyldithiocarbamat unverzüglich hinzugefügt. Dann wird 5 Minuten lang energisch gerührt.
Das schwarze Gemisch wird in einem Scheidetrichter solange mit kleinen Portionen von Chloroform extrahiert, bis die
Chloroformextrakte nur noch schwach gefärbt sind. Nach jedem Ausschütteln wird die schwerere Chloroformphase in einen
Erlenmeyerkolben abgelassen. Anfangs muss Geduld aufgebracht werden, da die Grenzfläche zwischen den Phasen nicht gut
zu erkennen ist. Die gesammelten Chloroformextrakte werden im Erlenmeyerkolben mit wasserfreiem Magnesiumsulfat versetzt
(ein paar Spatelspitzen) und zur Trocknung über Nacht mit einem Stopfen verschlossen stehen gelassen. Dann wird abfiltriert
und am Rotavapor auf ein Volumen von etwa 5 mL eingeengt. Dann fügt man langsam 25 mL Petrolether zu, worauf langsam schwarze
Kristalle wachsen (besser über Nacht gut verschlossen stehen lassen). Die Kristalle werden abfiltriert und an der HV getrocknet.
Nehmen Sie ein IR-Spektrum des Komplexes auf und ordnen Sie die Banden zu, die durch die NO-Gruppe verursacht werden. Freies NO absorbiert bei 1878 cm–1.
Das IR-Spektrum von Nitrosylbis(diethyldithiocarbamato)eisen:
-komplex_ir_spektrum.png)
Peakangaben zum IR-Spektrum von Nitrosylbis(diethyldithiocarbamato)eisen:
-komplex_ir_peaks.png)
Diskutieren Sie die möglichen Oxidationszustände von Eisen im Komplex. Stichwort: Enemark-Feldham-Notation.
Literatur: Synthese von [Co(NH3)5Cl]Cl2: G. G. Schlesinger, Inorg. Synth. 1967, 9, 160--163. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek Chemie: VH 5500; Synthese der Bindungsisomere: D. T. Richens, C. Glidewell in Inorganic Experiments, Wiley VCH, Weinheim, 2003, Seite 31--32. Nicht elektronisch verfügbar. Standortsignatur der Fachbibliothek Chemie: VH 5100 W913(2).
Benötigte Chemikalien: Ammoniumchlorid, 25%ige Ammoniaklösung, Cobalt(II)-chlorid Hexahydrat, 30% Wasserstoffperoxidlösung, konzentrierte Salzsäure, Wasser, Ethanol zum Waschen, Aceton zum Waschen, verdünnte Salzsäure, Natriumnitrit, Diethylether zum Waschen, Phosphorpentaoxid.
Darstellung von [Co(NH3)5Cl]Cl2
2,5 g Ammoniumchlorid werden in einem großen Erlenmeyerkolben in 16,3 mL 25%iger wässriger Ammoniaklösung gelöst. Unter ständigem Rühren
werden 5,0 g feingepulvertes Cobalt(II)-chlorid Hexahydrat in kleinen Portionen zugegeben. Die nächste Portion immer erst zugeben,
wenn sich die vorige aufgelöst hat. Unter Hitzeentwicklung bildet sich Hexammincobalt(II)-chlorid. Zur warmen Mischung gibt man
unter beständigem Rühren aus einer Bürette langsam, in einem dünnen Strahl 4,0 mL 30% Wasserstoffperoxidlösung. Die Reaktion verläuft
stark exotherm unter Sprudeln. Sobald das Sprudeln aufgehört hat, gibt man zur tiefroten Lösung langsam 15 mL konzentrierte
Salzsäure. Die Lösung wird auf dem Ölbad 15 Minuten lang auf 60 °C erhitzt (Gummistopfen auf Erlenmeyerkolben legen),
anschließend abgekühlt auf Raumtemperatur und abgenutscht.
Der Niederschlag wird zuerst mit kleinen Portionen von eiskaltem Wasser gewaschen (insgesamt 10 mL), dann mit 10 mL
halbkonzentrierter Salzsäure. Darauf wird zuerst mit Ethanol gewaschen, dann mit Aceton, und anschließend wird der Niederschlag
auf der Nutsche gründlich trocken gesaugt. Das auf diese Weise erhaltene Produkt ist in der Regel rein genug,
um damit weiterzuarbeiten.
Nur falls das UV-Spektrum dieses Produkts nicht dem unten abgebildeten entspricht, nehmen Sie folgenden Reinigungsschritt vor:
Das Produkt wird unter leichtem Erwärmen in etwa 45 mL 1 mol/l wässriger Ammoniaklösung gelöst und dann (unbedint im Abzug!)
in 45 mL konzentrierte Salzsäure gegossen. Es wird 45 Minuten auf dem Ölbad erhitzt. Dann wird das Komplexsalz wie oben
beschrieben isoliert, gewaschen und getrocknet.
Wiegen Sie die erhaltene Menge an Produkt und verteilen Sie sie 1:1 auf die beiden Synthesen der Bindungsisomere. Nachfolgend
werden die Synthesen basierend auf 1,5 g [Co(NH3)5Cl]Cl2 beschrieben. Passen Sie entsprechend
Ihrer Ausbeute an!
Darstellung von Bindungsisomer 1
1,5 g [Co(NH3)5Cl]Cl2 werden in einer Lösung aus 15 mL Wasser und 5 mL von 6 mol/L Ammoniak gelöst. Dazu wird unter Rühren erwärmt (auch kurzes Aufkochen kann nötig sein). Dann filtriert man, lässt abkühlen und säuert mit verdünnter Salzsäure an (etwa 5--10 mL). Dann werden 2,0 g Natriumnitrit zugegeben und vorsichtig erwärmt, bis sich der anfangs gebildete rote Niederschlag wieder aufgelöst hat. Man lässt abkühlen und fügt dann vorsichtig 20 mL konzentrierte Salzsäure zu. Kommt es zu merklichem Sprudeln, wird die Lösung in Eis gekühlt, worauf dann die braun-gelben Kristalle abfiltriert werden. Man wäscht zuerst mit Ethanol, dann mit Diethylether und trocknet dann die Kristalle im Exsikator über Phosphorpentaoxid.
Darstellung von Bindungsisomer 2
1,5 g [Co(NH3)5Cl]Cl2 werden in einer Lösung aus 25 mL Wasser und 5 mL konzentriertem Ammoniak gelöst. Dazu wird unter Rühren erwärmt (auch kurzes Aufkochen kann nötig sein). Es wird abfiltriert und dann dem Filtrat tropfenweise halbkonzentrierte Salzsäure zugefügt (etwa 15 mL) bis die Lösung gegenüber Universalindikatorpapier gerade neutral ist. Achtung: Die Lösung darf nicht alkalisch sein! Zur kalten Lösung werden dann 1,5 g Natriumnitrit und 1,5 mL halbkonzentrierte Salzsäure gegeben und lässt 1--2 Stunden in Eis stehen. Dann nutscht man den lachsfarbenen Niederschlag aus der eiskalten Lösung so schnell wie möglich ab, wäscht mit Eiswasser gefolgt von Ethanol gefolgt von Diethylether und trocknet ihn dann im Exsikator über Phosphorpentaoxid.
Nehmen Sie ein UV-Spektrum von einer verdünnten wässrigen Lösung von [Co(NH3)5Cl]Cl2 auf und vergleichen Sie es mit dem unten abgebildeten. Entspricht es diesem, fahren Sie fort mit der Synthese der beiden Bindungsisomere, ansonsten nehmen Sie den oben beschriebenen Reinigungsschritt vor.
Das UV-Spektrum einer wässrigen Lösung von [Co(NH3)5Cl]Cl2:
5cl]cl2_uv.png)
Das UV-Spektrum zeigt bei der langwelligen Bande eine deutliche Schulter. Versuchen Sie sie zu erklären.
Nehmen Sie von beiden Isomeren ein IR-Spektrum auf und ordnen Sie die Banden zu, die durch die NO2-Gruppe bzw. die ONO-Gruppe verursacht werden. Die Spektren sollten den beiden nachfolgend abgebildeten entsprechen.
Das IR-Spektrum von Isomer 1:
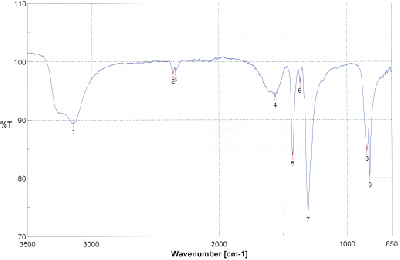
Peakangaben zum IR-Spektrum von Isomer 1:

Das IR-Spektrum von Isomer 2:
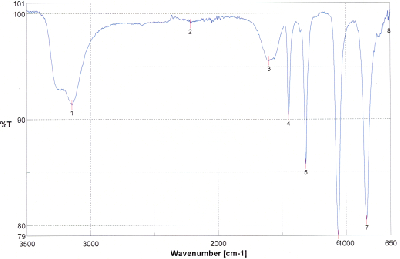
Peakangaben zum IR-Spektrum von Isomer 2:

Setzen Sie kleine Mengen ihrer beiden Isomere a) im Ofen der Hitze aus und b) mehrere Tage dem Sonnenlicht. Was passiert? Welches Isomer ist thermodynamisch stabiler?