Beispiel 1: Na2[Fe(C4H6O3)2] · 4 H2O in der Raumgruppe P 1
Sie lernen bei diesem Beispiel das trikline Kristallsystem mit seinen beiden Raumgruppen kennen, lösen eine Struktur durch trial and error und Differenzfouriersynthese, verfeinern mit der Kleinste-Fehler-Quadrate-Methode, führen Wasserstoffatome ein, stellen das Ergebnis graphisch dar. (Noch nichts wird gesagt über: Kristallauswahl und Messung, Gitterpunkte, Gitter vs. Struktur, Umgang mit Symmetrie außer 1, Strukturlösung mit Direkten oder Vektormethoden).
Trial and error. Im folgenden wird der Ablauf einer Strukturanalyse an einem Beispiel beschrieben, bei dem nichts schiefgeht. Die Elementarzelle ist dabei so klein, daß wir die billigste aller „Methoden“ anwenden können, nämlich trial and error. Es ist nicht Standard, aber auch nicht abwegig so vorzugehen, da die meist eingesetzten Direkten Methoden bei kleinen Elementarzellen seltsam schwach erscheinen („klein“ heißt bei Molekülstrukturen meistens, daß nur 1 Schweratom in der Elementarzelle enthalten ist). Wenn Sie nach dem übernächsten Beispiel Direkte Methoden anwenden können, versuchen Sie es einmal bei diesem Datensatz — Sie werden überrascht sein, was Sie sowohl von SHELXS als auch von SIR97 vorgesetzt bekommen (auch wenn Sie es mit Vektormethoden, auf die noch später eingegangen wird, versuchen, wird es nicht besser).
Der Kristall. Die untersuchte Substanz ist Na2[Fe(C4H6O3)2] · 4 H2O und wurde von H. Piotrowski in Form roter Kristalle aus blauen, wäßrig-natronalkalischen Lösungen isoliert, die neben Eisen(ii)-chlorid einen reichlichen Überschuß an Anhydroerythrit (C4H8O3, meso-Oxolan-3,4-diol) enthielten. Beabsichtigt war die Synthese eines Komplexanions, in dem Eisen(ii) an doppelt deprotoniertes Anhydroerythrit (meso-Oxolan-3,4-diolat(2–)) als Chelatliganden gebunden sein sollte, vielleicht neben Aqua- oder Hydroxo-Liganden. Die Elementaranalyse zeigt, daß pro mol Eisen 2 mol Natrium, 2 mol Oxolandiolat und 4 mol Wasser enthalten ist. Die Summenformel wäre dann C8H20FeNa2O10. Die Dichte der roten Kristalle wurde nicht bestimmt.
Die Metrik. Wir beginnen die Strukturanalyse mit einem Blick auf die Abmessungen der Elementarzelle, der „Metrik“. Das Diffraktometer gibt die folgenden sechs Werte aus (die Achslängen a, b, c sind in Å [Ångstrom-Einheiten, 1 Å = 10–10 m], die Winkel α, β, γ in Grad angegeben):
a = 5.2479(10), b = 8.4160(16), c = 9.1386(18), α = 109.035(19), β = 97.73(2), γ = 106.978(19)
Standardabweichungen. Die Zahlen in Klammern sind, wie im folgenden stets, die Standardabweichung (σ) der letzten Dezimalstelle. 5.2479(10) bedeutet 5.2479 mit σ = 0.0010. In der Literatur sind zwei Schemata üblich. Beim ersten werden der letzten Dezimalstelle σ-Werte zwischen 1 und 9 zugeordnet, was zu einem „harten“ Übergang von 9 nach 1 führt. In diesem Kurs wird einheitlich ein zweites Schema mit Werten zwischen 1 und 19 angewendet. Die Diffraktometerausgabe für den Winkel β von 97.734(20) wird also zu 97.73(2) gerundet, während α als 109.035(19) angegeben ist.
Kristallsystem, Kristallklasse. Zwischen den sechs Gitterkonstanten sind keine besonderen Zahlenbeziehungen erkennbar, etwa daß zwei Achslängen fast gleich wären oder daß zwei oder drei Winkel fast gleich oder nahe bei 90 oder 120� wären. „Fast“ und „nahe bei“ bedeutet „gleich“ innerhalb 2 bis 3 σ. Die Ursache für eine Metrik mit sechs unabhängigen Gitterkonstanten ist, daß in der Kristallstruktur nur die beiden Symmetrieelemente 1 oder 1 (sprich: eins-quer) vorkommen. Wir verwenden für Symmetriesymbole die Nomenklatur nach Hermann-Maugin; 1 und 1 heißen bei Schoenflies E und i, es handelt sich also um die Identität (vulgo: „keine“ Symmetrie) und die Inversion (Punktspiegelung, in der Kristallographie heißt 1 meist „Symmetriezentrum“ oder kurz „Zentrum“). Kommen in einer Kristallstruktur keine weiteren Symmetrielemente vor, so gehört sie zum triklinen (deutsch: dreifach geneigten, früher auch: „anorthischen“, deutsch: frei von rechten Winkeln) Kristallsystem. Das trikline Kristallsystem zerfällt in die beiden Kristallklassen 1 und 1, worunter man die nach außen sichtbare Symmetrie des Kristalls versteht, die man durch ein Punktgruppensymbol beschreibt. Nach Hermann-Maugin haben Symmetrieelemente und Punktgruppen in diesen beiden Fällen dieselbe Bezeichnung (bei Schoenflies ist das anders, hier würde man die Kristallklassen mit C1 und Ci bezeichnen).
Elementarzelle, Raumgruppe, asymmetrische Einheit. Um die Struktur, also die Anordnung der Atome im Raum, zu beschreiben, nutzen wir die Vorstellung, daß in einem unendlich ausgedehnten Muster Translationssymmetrie möglich ist, daß wir also die Atomanordnung als dreidimensional wiederkehrende Anordnung eines Basismotivs verstehen können. Im Verlauf einer Strukturanalyse ermitteln wir nun dieses Basismotiv. Der diesem Motiv zugeordnete Raum ist die Elementarzelle, die durch drei Vektoren in die drei Raumrichtungen vervielfältigt wird. Die drei Vektoren sowie deren relative Richtung im Raum, also die Winkel zwischen ihnen, haben wir bereits kennengelernt, nämlich die sechs Gitterkonstanten, die die Elementarzelle aufspannen. Der nächste Schritt besteht darin, die Elementarzelle mit Atomen zu füllen, vorher sehen wir uns aber noch an, welche Symmetrieeigenschaften der zu füllende „Kasten“ hat. Diese sind in einem Symbol für die jeweilige Raumgruppe zusammengefaßt. In jeder Kristallklasse des triklinen Kristallsystems gibt es genau 1 Raumgruppe, und zwar P 1 in der Kristallklasse 1 und P 1 in der Kristallklasse 1. Erneut wiederholen sich die Symmetrieangaben: Liegt eine Struktur in der Raumgruppe P 1 vor, so gibt es außer der Identität keine weitere Symmetrie, so daß wir die gesamte Elementarzelle mit Atomen zu füllen haben. Anders in der Raumgruppe P 1. Hier wird jedes Molekülfragment durch ein Symmetriezentrum in ein gleich großes, invertiertes Bildobjekt überführt. Es bleibt also nur das halbe Zellvolumen zur Füllung mit symmetrisch unabhängigen Atomen — die asymmetrische Einheit ist halb so groß wie die Elementarzelle. Hat das Molekül selbst die Punktsymmetrie 1, so muß bei der Strukturanalyse nur eine Hälfte festgelegt werden, falls das Symmetriezentrum des Moleküls und das Symmetriezentrum der Elementarzelle zusammenfallen (das muß nicht sein, aber meistens ist es so; Molekülsymmetrie, die nicht deckungsgleich mit der Symmetrie der Packung im Kristall ist, nennt man „nichtkristallographische Symmetrie“, auf die später eingegangen wird). Was bedeutet nun noch das „P“ im Raumgruppensymbol? Es besagt, daß die Elementarzelle „primitiv“ ist, was bedeutet, daß keine anderen Vektoren als die drei Gitterkonstanten a, b und c zur Translation des Basismotivs benutzt werden. In Matrixschreibweise: nur die Vektoren [100], [010] und [001] sowie deren Kombinationen repräsentieren die Translationssymmetrie der Struktur. (Hier liegt ein sprachlicher Fallstrick für den Anfänger: Wie später gezeigt wird, gibt es außer den primitiven auch „zentrierte“ Zellen. Hat eine Elementarzelle — egal ob primitiv oder zentriert — ein Symmetriezentrum, so heißt sie „zentrosymmetrisch“ oder kurz: „zentrisch“. P 1 ist also zentrisch, aber nicht zentriert! In Publikationen auf Englisch ist es nicht besser: P 1 is centric but not centered!)
Zahl der Formeleinheiten. Versuchen wir nun also, die Elementarzelle in unserem Beispiel zu füllen. Erste Frage: Wieviele Atome müssen eigentlich hinein? Dies ist die Frage nach der Zahl der Formeleinheiten in der Elementarzelle, üblicherweise mit Z abgekürzt. Z kann leicht ausgerechnet werden, wenn (a) die Summenformel und damit auch die Formelmasse, (b) das Volumen der Elementarzelle und (c) die Dichte der Substanz bekannt ist. Eine hinreichend genaue Summenformel kann durch Analyse erhalten werden; das Volumen der Elementarzelle kann aus den Gitterkonstanten errechnet werden und ist gewöhnlich dem Meßprotokoll des Diffraktometers zu entnehmen. Dichtebestimmungen sind allerdings ziemlich aus der Mode gekommen. Für die Praxis läßt sich dies bei Molekülverbindungen meist verschmerzen, da sich die ganze Prozedur durch eine einfache Faustregel näherungsweise ersetzen läßt:
Jedes Nichtwasserstoffatom trägt mit ungefähr 19 Å3 zum Elementarzellvolumen bei.
In unserem Beispiel beträgt das Zellvolumen gerundet 393 Å3. Dividiert durch 19 ergeben sich 20.7 Atome pro Elementarzelle. Addieren wir die Nichtwasserstoffatome in der Summenformel C8H20FeNa2O10, so erhalten wir 8 + 1 + 2 + 10 = 21. Damit ist die Zahl der Formeleinheiten in der Zelle klar: Z = 1, in der ganzen Elementarzelle ist also nur eine einzige Formeleinheit — und damit nur ein einziges Eisenatom. Warum ist dies wichtig? Röntgenstrahlung wechselwirkt mit Elektronen. Je elektronenreicher ein Atom, umso höher sein Beitrag zum Beugungsmuster. Eine Röntgenstrukturanalyse geht daher im Gegensatz zu einer Neutronenstrukturanalyse so vor sich, daß zuerst die Lagen der Schweratome bestimmt werden, um in anschließenden Differenzfouriersynthesen zunehmend leichtere Atome zu lokalisieren.
Wir legen die Schweratomlage fest. Aber der Reihe nach: Um dies zu tun, müssen wir uns für eine Raumgruppe entscheiden. Und nun kommt der Schuß ins Blaue (vornehmer: trial and error): Keines der Edukte ist chiral. Damit ist die Wahrscheinlichkeit recht hoch, daß die Struktur zentrosymmetrisch ist (ist wenigstens ein Edukt chiral, ist es genau umgekehrt: die Struktur kann dann nicht zentrosymmtrisch sein, da das Symmetrielement 1 von jedem Baustein ein Spiegelbild erzeugt; wird in eine Lösung aber zum Beispiel nur l-Histidin hineingegeben, dann fehlt d-Histidin, um mit seinem Enantiomer einen zentrosymmetrischen Kristall aufzubauen). Nehmen wir also an, daß unsere Struktur in der Raumgruppe P 1 zu beschreiben ist, können wir sofort die Lage des schwersten Atoms angeben! Da nämlich nur 1 Eisenatom pro Elementarzelle enthalten ist, muß dieses genau auf einem Symmetriezentrum liegen. Wäre dies nicht der Fall, würde das Symmetriezentrum ein zweites Eisenatom erzeugen, was aber im Widerspruch zur Bestimmung von Z wäre! Zur Sprechweise: Das Eisenatom kann nicht die allgemeine, sondern nur eine spezielle Punktlage besetzen.
Die Internationalen Tabellen: Alles über Raumgruppen. Wo liegen aber Symmetriezentren in P 1? Hierüber geben die International Tables for X-Ray Crystallography, Vol. 1, Auskunft. Die Raumgruppe P 1 ist als Nummer 2 aufgeführt. Auf der rechten Seite ist eine Liste der Punktlagen (“Positions”) zu sehen. Wie bei jeder Raumgruppe beginnt die Aufzählung mit der allgemeinen Punktlage, deren Zeile wie folgt aussieht:
2 i 1 (1) x, y, z (2) x, y, z.
Die erste Ziffer ist die „Zähligkeit“ (“Multiplicity”) der Punktlage. Sie gibt an, wieviel Objekte durch Symmetrie insgesamt erzeugt werden, wenn auf diese Punktlage 1 Objekt gelegt wird. Hier also: 1 Originalobjekt + das durch 1 invertierte Bildobjekt = 2 Objekte insgesamt. Der nächste Eintrag, das Wyckoff-Symbol i ermöglicht eine eindeutige Kurzbezeichnung der Punktlage (indem man Zähligkeit und Wyckoff-Symbol zusammenzieht; wir reden also im Augenblick von der Punktlage 2i). Der dritte Eintrag nennt die Lagesymmetrie (“Site symmetry”), die lokale Symmetrie eines Objektes auf dieser Lage, die bei der allgemeinen Punktlage in jeder Raumgruppe immer 1 ist. Die anschließend angegeben Koordinaten wiederholen auf ihre Art das bisher gesagte: Legen wir ein Atom auf irgend einen Punkt mit den Achsabschnitten x, y, z in dem durch die Vektoren a, b und c aufgespannten Koordinatensystem (außer auf die dann folgenden speziellen Punktlagen natürlich), dann erzeugt ein Symmetriezentrum (das selbst offensichtlich die Koordinaten 0, 0, 0 aufweist) ein zweites Atom mit den Koordinaten –x, –y, –z (man beachte in den Tabellen den Kristallographenbrauch, das Minuszeichen als Überstrich zu schreiben). Wird ein Baumotiv, zum Beispiel ein Atom, auf eine der anschließend aufgelisteten speziellen Punktlagen gelegt, so ist dort die Zähligkeit immer kleiner als in allgemeiner Lage. Bei allen weiteren Einträgen ist die Zähligkeit 1 und die Lagesymmetrie 1. Auf welches der acht angegebenen Symmetriezentren ist nun das Eisenatom zu legen? Es ist egal. Beim ersten Atom, das festgelegt wird (und nur dort!), sind in P 1 alle speziellen Lagen äquivalent und können durch Ursprungsverschiebung ineinander überführt werden. Um die Elementarzelle übersichtlich zu füllen, wählen wir einfach deren Mittelpunkt, also die Punktlage 1h mit den Koordinaten ½, ½, ½ als Lage für das Eisenatom.
Rechnen mit SHELXL. Der nächste Schritt besteht in einer Differenzfouriersynthese, die die Lagen der ersten leichteren Atome ergeben soll. Als Hilfsmittel dient SHELXL, das für die Berechnung zwei Dateien erwartet, die vom Typ name.hkl und name.ins sind. name ist in diesem ersten Beispiel b14.
b14.hkl
enthält die Beugungsintensitäten; sie steht als Diffraktometerausgabe zur Verfügung. Jede Zeile enthält h, k, l, Fo2 und σ(Fo2).
b14.ins
ist eine instruction-Datei mit Anweisungen, was SHELXL tun soll. Nach der obligatorischen TITL-Anweisung, die ab Spalte 5 beliebigen Text enthalten darf, folgen CELL, ZERR, SFAC und UNIT; alle weiteren Anweisungen können in beliebiger Reihenfolge eingefügt werden mit Ausnahme der HKLF-Anweisung, die als letzte erwartet wird (daran anschließender Text wird überlesen). Im einzelnen:
TITL irgend ein Text
CELL Wellenlänge der Röntgenstrahlung, a, b, c in Å, α, β, γ
ZERR Z, σ(a), etc.
SFAC Atomsorten
UNIT Zahl der Atome jeder Sorte in der Elementarzelle
FMAP Steuerzahl 2 für Differenzfouriersynthese
PLAN Zahl der zu berücksichtigenden Fouriermaxima
Atom SFAC-Nummer, x, y, z, (10 +) k, U
HKLF Steuerzahl 4 für F2-basierte Beugungsdaten
Die Reihenfolge der Elemente auf der SFAC-Karte ist frei, es schadet aber nicht, der Chemical-Abstracts-Reihenfolge für Summenformeln zu folgen (zuerst C und H, dann den Rest in alphabetischer Reihenfolge), da manche Zeitschriften dies verlangen. Bei der PLAN-Anweisung ist die Zahl der zu berücksichtigenden Fouriermaxima mit –1 multipliziert, wodurch die neu gefundenen Elektronendichten mit den schon vorhandenen Atomen zu Baueinheiten kombiniert werden. Auf der Atom-Anweisung bedeutet k den Besetzungsfaktor. Bei einer voll besetzten Lage ist dies der Quotient aus der Zähligkeit der betreffenden Lage und der Zähligkeit der allgemeinen Lage. Bei der speziellen Punktlage ½, ½, ½ ist k also 1:2 = 0.5. Soll ein k-Wert nicht als freie Variable verfeinert werden, so wird 10 addiert, daher der Wert 10.5. (Die programmtechnische Behandlung spezieller Punktlagen zeigt einen anderen gedanklichen Zugang zu diesen: Jeder Baustein wird durch die Zentrosymmetrie verdoppelt, egal wo er liegt. Auf der speziellen Lage liegt nun aber kein Atom, das besonders behandelt wird, sondern nur ein halbes, das durch Anwenden der Symmetriebedingung zu einem ganzen Atom ergänzt wird.) U ist der isotrope Temperaturparameter in Å2, für den ein Schätzwert eingegeben ist.
Differenzfouriersynthese. Durch Fourier-Transformation läßt sich aus einer Struktur, also einer vollständig besetzten Elementarzelle, das Beugungsdiagramm berechnen. Jeder Röntgenreflex ist dabei durch seine Lage im Raum (festgelegt durch die Metrik) sowie durch seinen Strukturfaktor charakterisiert, der Betrag und Phase aufweist. Die umgekehrte Fourier-Transformation würde die Strukturfaktoren in die Struktur überführen. Leider gibt das Diffraktometer keine Strukturfaktoren aus, sondern nur Beugungsintensitäten, die dem Quadrat der Strukturfaktoren proportional sind. Es ergibt sich das bekannte „Phasenproblem“ der Röntgenkristallographie. Im Fall einer zentrosymmetrischen Struktur läßt sich das Problem anschaulich darstellen. Hier sind nur zwei Werte für die Phase möglich: 0 und 180� entsprechend einem positiven oder negativen Vorzeichen des Strukturfaktors F. Da die Intensität der gebeugten Strahlung proportional zu F2 ist, ergibt sich unabhängig vom Vorzeichen des Strukturfaktors stets ein positiver Wert für F2. Strukturlösen heißt also, allen Strukturfaktoren das aus der Messung nicht zugängliche, korrekte Vorzeichen zuzuordnen. Ist dies gelungen, ergibt eine Fourier-Tranformation eindeutig die Elektronendichteverteilung, also die Struktur.
Welches Ergebnis hätte eine Fourier-Transformation bei unserer Beispielstruktur, wenn nicht alle Atome der Elementarzelle in die Berechnung einbezogen würden, sondern wenn zum Beispiel alle Wassermoleküle weggelassen würden, alle anderen Atome aber, vor allem das einzige Schweratom Eisen, am richtigen Platz lägen? Da der Eingriff nicht sehr drastisch ist, werden alle Strukturfaktoren um einen gewissen Betrag verändert sein: aus +100 würde vielleicht +90, aus –250 vielleicht –240, aus +10 vielleicht –10. Man beachte, daß bei den starken Reflexen mit einem hohen Betrag des Strukturfaktors nicht mit einer Vorzeichenänderung zu rechnen ist! Hieraus ergibt sich das Prinzip der Fouriersynthese: Werden die Vorzeichen der berechneten F-Werte (Fc, der Index kommt von “calculated”) einer unvollständig gelösten Struktur den experimentell bestimmten Beträgen der Strukturfaktoren (|Fo|, der Index kommt von “observed”) zugeordnet, dann sollte eine Fourier-Transformation dort Elektronendichte ergeben, wo noch nicht lokalisierte Atome liegen. Die Praxis zeigt, daß ein besseres Ergebnis bei einer Differenzfouriersynthese erhalten wird: Hier werden die Vorzeichen nicht mit |Fo| kombiniert, sondern mit |Fo| – |Fc|, wobei nur der noch nicht erkannte Teil der Struktur in der Fourier-Transformation sichtbar wird.
b14.lst und b14.res. Sehen wir uns aber die gesamte Ausgabedatei
b14.lst
an. SHELXL mittelt über symmetriegleiche Reflexe. In der triklinen Raumgruppe P 1 wird lediglich über Friedel-Paare gemittelt, das sind Paare h, k, l und –h, –k, –l sowie über mehrfach gemessene Reflexe. Aus 3525 Reflexen in b14.hkl werden so 1554 symmetrieunabhängige Reflexe (“unique reflections”). Der innere R-Wert R(int) von 0.0369 zeigt an, daß die Mittelung wirklich über ziemlich gleiche Reflexe ausgeführt wurde, daß also der Datensatz nicht in irgend einer Weise beschädigt ist (wenn wir in späteren Beispielen mehr Symmetrie zulassen, werden wir R(int) als Kontrollparameter kennenlernen, ob zuviel Symmetrie angenommen wurde).
Bei der abschließenden Strukturfaktorberechnung werden weitere R-Werte ausgegeben. wR2 ist der gewichtete R-Wert über F2, R1 der „konventionelle“ R-Wert, das ist der ungewichtete R-Wert über F. In diesem sehr frühen Stadium der Strukturbestimmung gibt R1 ein vages Gefühl, ob alles o.k. ist (es ist; 0.46 ist ordentlich für so wenig korrekt beschriebene Elektronendichte; wäre die Schweratomlage falsch gewesen, wäre ein Wert über vielleicht 0.8 herausgekommen).
Unter dem Sternenhimmel aus Ziffern dann die Differenzfouriersynthese. Nach dem Eisenatom beginnt jedes Restdichtemaximum mit einer Laufnummer, dann folgt die Restdichte in e Å–3, dann x, y, z und k (dann kommt etwas ganz Tolles, was heute niemand mehr braucht, nämlich die Lage des Atoms in cm über dem Papier; man kann nämlich eine Eisenplatte unter den Ausdruck legen und darauf dann Magnete mit Spießen daran auf die Ziffern des Sternenhimmels stellen und schließlich einen bunten Tischtennisball in eben dieser cm-Höhe auf den Spieß stecken und dann sieht man die Struktur. Ja, früher ...). Auf die Tabellen mit Abständen und Winkeln gehen wir später ein. Blättert man nun durch den Text, so sieht man die Restdichten allmählich kleiner werden — bis zu einem Sprung nach dem Restmaximum mit der Laufnummer 10. Versuche, Atome zuzuordnen, sollten bei einem solchen Sprung enden. Um nun möglichst angenehm fortzufahren, benutzen wir die Hilfe eines Graphikprogramms wie zum Beispiel SCHAKAL. Als Eingabedatei verlangt SCHAKAL eine Datei, die von SHELXL geschrieben wird und die kürzer als b14.lst das Wesentliche zusammenfaßt, und zwar die Datei
b14.res
. Die results-Datei sieht so aus wie eine ins-Datei. Die Restdichten sind durch das „Atomsymbol“ Q gekennzeichnet. Der Abbruch nach Restdichte 10 wird in dieser Liste gut deutlich.
SCHAKAL. SCHAKAL kennt keine Atome, die „Q“ heißen. Man benennt daher die Qs in irgend etwas um, zum Beispiel in F und kopiert die Restmaxima, die Atome sein könnten, vor die abschließende HKLF-Anweisung (auch SCHAKAL behandelt die HKLF-Anweisung als Datei-Ende-Marke). Anschließend kopiert man die so veränderte Datei in das Verzeichnis schakalordner/dat. schakalordner ist der Ordner, in dem sich auf Ihrem Rechner die SCHAKAL-Programmdateien befinden (jetzt und im Folgenden wird die auch bei HTML-Verweisen übliche UNIX-Schreibweise für Verzeichnisse beibehalten; auf einem Windows-Rechner wäre also schakalordner/dat durch schakalordner\dat zu ersetzen). Man startet SCHAKAL und gibt ein:
u x (use shelx file) es wird nach dem Namen der res-Datei gefragt, hier: b14, und nach einem Namen für eine Datei für die SCHAKAL-Daten; es spricht nichts dagegen, noch einmal b14 einzugeben, es wird dann schakalordner/b14.dat angelegt. x zeichnet die Struktur; um sie passend hinzulegen, gibt man ein: o, man kann das Bild jetzt mit den Pfeiltasten drehen wie man will; wenn man fertig ist, nochmal x; jetzt fügt man die Atomnamen mit w a (write atoms) hinzu und schon sieht man das beinahe
fertige Bild
(beinahe, weil die gestrichelten Bindungen noch zu machen wären, was aber entbehrlich ist (wer unbedingt will: d g 1 [define group] ordnet vorsichtshalber alle Atome einer Gruppe 1 zu; d g 2 f1 f4 f5 definiert eine Atomgruppe 2 aus den eingegebenen drei Atomen; a b #'2=# < 3 [add bond] fügt alle Bindungen zwischen Atomen der Gruppe 2 und allen Atomen zu, solange sie kürzer als 3 Å sind; c f 7 #=# > 2.2 [change fragmentation] zerhackt Bindungen von allen zu allen Atomen in 7 Teile, wenn sie länger als 2.2 Å sind; größer 2.2 Å deshalb, damit nicht sämtliche Bindungen zerhackt werden]).
Differenzfouriermaxima zu Atomen zuordnen. Nun zur Interpretation der Differenzfouriersynthese, zu der Sie am Besten die lst-Datei und das SCHAKAL-Bild öffnen: Q1 bindet im Bild nur an Q2. Schauen wir nun in der lst-Datei unter den zu Q1 gehörenden Abständen nach, finden wir allerdings keinen Kontakt zu Q2. Vielleicht ist der Abstand so groß, daß SHELXL ihn nicht mehr als Bindung interpretiert. Werfen wir zur Sicherheit einen Blick auf die Abstände um Q2 ..., nanu! Hier gibt es einen Abstand von 2.427 Å zu Q1! Gleich beim ersten Beispiel zeigt sich hier eine notorische Schwäche von SHELXL: die Liste der Abstandswerte nach einer Fouriersynthese neigt zur Unvollständigkeit! Nun gut, merken wir uns diesen Abstand erst einmal und schauen wieder auf die Liste von Q1. Weitere Abstände zu Qs mit einer Laufnummer bis 10 finden sich am Ende der Liste und zwar drei Abstände knapp unter 2.4 Å zu den Restmaxima Q3, Q5 und Q4. Vor den Laufnummern der Q-Werte stehen weitere Zahlen, nämlich 16, 26 und 17. Die erste der drei Zeilen beginnt also mit „16 3“. Dies bedeutet, daß Q1 nicht etwa einen Kontakt zu Q3 aufweist, wenn das Q3 so, wie es in der res-Datei steht, betrachtet wird, sondern es ist zuerst eine Symmetrieoperation auf Q3 anzuwenden. Welche dies ist, steht am Schluß der Abstandstabellen für die Atome und Restmaxima eines Moleküls. Der Eintrag „16 3“ ist hier ergänzt durch die drei Werte x, y und z des symmetrietransformierten Q3 und es ist außerdem die Art der Symmetrieoperation angegeben, hier: 0.0000-X 1.0000-Y 1.0000-Z, also –x, 1 – y, 1 – z. Dies erklärt, warum die entsprechende Bindung nicht im SCHAKAL-Bild zu sehen ist, das nur mit den untransformierten Atomen der res-Datei gezeichnet worden ist. Wozu man das alles nutzen kann, sehen wir später. Die aufgelisteten Abstände um Q1 — ob vollständig oder nicht — genügen, diese größte Restdichte zu identifizieren. Es handelt sich um typische Na-O-Bindungen. Da Natrium nach Eisen der stärkste Streuer im Beispielkristall ist, fällt die Zuordnung von Q1 zu Natrium leicht. Bevor die restlichen Maxima zugeordnet werden, noch ein Blick auf die Einträge zu Q1: Nach der Höhe des Maximums von 15.84 e Å–3 folgen die Werte für x, y und z, anschließend folgt der Besetzungsparameter k. Der Wert von 1 zeigt, daß es sich um die allgemeine Punktlage handelt. Das Eisenatom dagegen liegt speziell mit k = 0.5, das heißt, in der Elementarzelle gibt es doppelt so viel Natrium wie Eisen. Dieser Befund ist beruhigend, denn in der Summenformel hieß es ...FeNa2...! Bevor wir nun die Umgebung eines jeden weiteren Restmaximums anhand der lst-Datei durchleuchten, schauen wir noch einmal auf das SCHAKAL-Bild. Eingesetzt wurde bei der Synthese der Substanz Anhydroerythrit (meso-Oxolandiol) — und genau das sieht man als Chelatligand an das Eisenatom binden. Daß das Eisenatom zweibindig zu sein scheint, macht nichts. Wie schon gesagt, wurde SCHAKAL nicht angewiesen, Symmetrie zu beachten. Wir aber wissen, daß das Eisenatom in spezieller Lage auf 1 liegt. Das Symmetriezentrum erzeugt zusätzlich zu dem jetzt sichtbaren Chelatligand einen zweiten, so daß für Eisen eine quadratisch-planare Umgebung resultiert (auch für die Atome des Oxolandiols ist k = 1 entsprechend zwei Liganden pro Eisenatom). Übrig bleiben nur noch die Restmaxima Q4 und Q5. Bei Q4 wiederholt sich der Ärger mit mit der SHELXL-Ausgabe: der gestrichelt eingezeichnete Kontakt zu Q2 ist bei Q4 nicht aufgelistet, wohl aber bei Q2; darüber hinaus bindet Q4 an ein symmetrieerzeugtes Natrium-Atom, es könnte sich also um ein Wasser-O-Atom handeln. Diese Vermutung wird durch den für eine Wasserstoffbrückenbindung typischen Abstand von 2.766 Å zu Q2 — einem Diol-O-Atom — gestützt. Überzeugen Sie sich davon, daß aus denselben Gründen auch Q5 ein Wasser-O-Atom sein sollte. Damit sind alle Restmaxima nennenswerter Höhe zugeordnet; das Ergebnis ist in einem
ergänzten SCHAKAL-Bild
zusammengefaßt und läßt sich in die
res-Datei
übertragen. Man beachte die Abfolge der Atome: nach dem Natriumatom folgen die O-Atome und schließlich die elektronenärmsten und daher streuschwächsten C-Atome. Zum Schluß eine kurze Bilanz, wie weit wir gekommen sind: Pro halbes Eisenatom wurden 1 Natriumatom, C- und O-Atome eines Oxolandiols sowie 2 Wasser-O-Atome lokalisiert. × 2 genommen erhält man den durch Elementaranalyse und Abschätzung von Z ermittelten Gesamtbesatz der Elementarzelle mit Nichtwasserstoffatomen! Ob wir fast fertig sind? Wenn das mal gut geht.
Kleinste-Fehlerquadrate-Verfeinerung, Wichtung. Der nächste Schritt wird vorbereitet, indem den neugefundenen Atomen die korrekten SFAC-Nummern zugewiesen werden, außerdem könnte man die Atomliste durch Kommentarkarten (REMark-Anweisung, die ab Spalte 5 beliebigen Text enthalten darf) etwas gliedern. Die wichtigste neue Anweisung in der res-Datei, die nach dem Editieren in
b14.ins
umbenannt wird, ist die die L.S.-Anweisung (Least Squares). Bei der Verfeinerung werden die Ausgangswerte von x, y und z sowie der Temperaturparameter so optimiert, daß wR2 minimal wird. L.S. 8 bedeutet, daß insgesamt 8 Verfeinerungscyclen durchlaufen werden sollen. Das „w“ in wR2 deutet darauf hin, daß es sich um einen gewichteten R-Wert handelt, bei dessen Berechnung jeder Reflex ein Gewicht hat, das möglichst nahe bei σ(F2)–2 liegt. Eine experimentelle Bestimmung von σ(F2), der Standardabweichung von F2, ist jedoch sehr unüblich (man müßte zum Beispiel zehnmal einen Datensatz auf dem Diffraktometer bestimmen um bei der Mittelwertbildung die gesuchte Standardabweichung zu erhalten) und man behilft sich daher mit einer Abschätzung aus der Zählstatistik. Hierbei kommen die starken Reflexe aber viel zu gut weg. SHELXL berechnet Gewichte nach ..., wobei die Parameter x und y (nicht mit den gleichnamigen Lageparametern verwechseln!) die Größe des Beitrags definieren, der zu σ(F2)–2 hinzuaddiert wird. Die beiden Gewichtsparameter werden gegen Ende der Verfeinerung optimiert. Jetzt ist es noch zu früh dazu, so daß grob mit x = 0.1 und y = 0 gerechnet wird. Dies ist der Inhalt der WGHT-Anweisung, die in der ins-Datei auf die L.S.-Anweisung folgt (man sieht dort nur den Wert für x, der fehlende y-Wert wird als 0 interpretiert).
Die mitverfeinerten Temperaturparameter sind auf dieser Stufe der Verfeinerung isotrop. Die „Verschmierung“ der Elektronendichte eines Atoms durch Temperaturbewegung wird also als gleich in allen Raumrichtungen angenähert, weshalb 1 Parameter reicht. Die Angabe ist in Å2. Ein typischer Wert von 0.04 Å2 entspricht also einer Schwingungsamplitude von 0.2 Å. Gegenüber der ersten ins-Datei ist schließlich noch die FVAR-Anweisung (Free VARiable) neu, die zur Zeit nur einen Wert enthält, den Skalierungsfaktor (overall scale factor), der bei jeder Strukturfaktorberechnung ungefragt angepaßt wird. Ferner wurde die Anweisung MOLE 1 eingefügt, die bewirkt, daß alle Atome nach der Differenzfouriersynthese in eine Liste gelangen. Läßt man SHELXL mit dieser ins-Datei starten, wird erneut
b14.lst
und
b14.res
geschrieben (wenn Sie unter Windows oder Unix Dateien zu Übungszwecken speichern wollen, müssen Sie diese vor weiteren SHELXL-Läufen umbenennen, da diese Betriebssysteme nicht wie zum Beispiel VMS Datei-Versionen speichert!) Sehen Sie sich in der lst-Datei den Fortschritt der Verfeinerung an. Nach dem letzten Cyclus wird der Maximalwert für den Quotient aus Verschiebung und geschätzter Standardabweichung (shift/esd = shift/estimated standard deviation) mit –0.001 angegeben, die Verfeinerung ist also zur Konvergenz gelangt. Wäre dies nicht so, würde man das Ganze mit der neuen res-Datei wiederholen und vielleicht außerdem die Zahl der Verfeinerungscyclen erhöhen. Die verfeinerten Temperaturparameter liegen alle nahe beieinander. In späteren Beispielen wird darauf eingegangen, was daraus zu lernen ist, sollte dies einmal nicht so sein. Es fehlt noch ein Blick auf die Maxima der neuen Differenzfouriersynthese: Ca. 1 e Å–3 ist zu wenig, um noch Nicht-H-Atome zu lokalisieren. Wir finden dementsprechend die höchsten Maxima nur 0.5 Å von O1 entfernt entsprechend einer ausgeprägten anisotropen Schwingung (O1 ist als Ether-O-Atom nur an 2 C-Atome gebunden und sollte senkrecht zu den beiden Bindungen weit ausschwingen können) und/oder wir sehen schon die freien Elektronenpaare des O-Atoms, für die ebenfalls der 0.5-Å-Abstand typisch ist. Kurz: Alles ist unauffällig, so daß der nächste Schritt getan werden kann. Wir ordnen allen Atomen anisotrope Temperaturparameter zu; außerdem fügen wir dort Wasserstoffatome an, wo sich deren Lage zwangsläufig ergibt, also bei den bislang zwei- und dreibindigen C-Atomen.
Wasserstoffatome in berechneten Positionen. Um an die C-Atome H-Atome anzufügen, stehen in SHELXL HFIX-Anweisungen zur Verfügung, die in die neue Version von
b14.ins
eingefügt werden. „HFIX 23 21 C1 C4“ bedeutet, daß an die Atome C1 und C4 je 2 H-Atome angefügt werden sollen, deren Lage sich ergibt, wenn die Umgebung dieser C-Atome zum Tetraeder ergänzt wird. Dei Steuerzahl 23 definiert Methylen-H-Atome (CH2). Die folgende Zahl 21 steht für den isotropen Temperaturfaktor, der den neuen Atomen zugewiesen werden soll. Da H-Lagen aus naturgesetzlichen Gründen mit hohem systematischen Fehler behaftet sind, hat es keinen Sinn, hohe Präzision vorzutäuschen, wo nichts sein kann. Eine Möglichkeit, sich nicht unnötig viele neue Variable durch H-Atome einzuhandeln, besteht darin, allen Wasserstoffatomen einen gemeinsamen Temperaturfaktor zuzuweisen. So etwas macht man in SHELXL mit der FVAR-Anweisung. „21“ liest sich so: nimm 1 mal die freie Variable 2. Der zugehörige Zahlenwert findet sich nun an zweiter Stelle auf der FVAR-Anweisung. 0.05 ist ein Schätzwert, von dem aus die Verfeinerung starten soll. Die zweite HFIX-Anweisung ist entsprechend aufgebaut; die Steuerzahl 13 definiert hier Methylidengruppen (CH). Es bleibt die Aufgabe, in der nächsten Differenzfouriersynthese die an Sauerstoff gebundenen H-Atome zu lokalisieren. Dies geht mal leichter und mal schwerer, wenn die O-Atome anisotrop verfeinert wurden (leichter, weil der wR2 besser und die Fouriersynthese damit weniger Artefakte enthält; schlechter, weil die durch Protonen bedingte Asymmetrie in der Elektronendichteverteilung um die O-Atome durch eine anisotrope Verfeinerung „glattgebügelt“ wird). Versuchen wir es mit der Variante „anisotrop vorher“. Hierzu genügt es, die Anweisung ANIS ohne Parameter einzufügen; die Anweisung wirkt dann auf alle Nicht-H-Atome. Nach dem SHELXL-Lauf gibt es dann wieder
b14.lst
und
b14.res
. Wieder wurde Konvergenz erreicht und auch dem wR2-Wert geht es zunehmend besser (das Ziel ist, ungefähr R(int) zu erreichen).
Wasserstoffatome in der Differenzfouriersynthese suchen. Was noch fehlt, sind zweimal 2 Wasserstoffatome, um aus O91 und O92 Wassermoleküle zu machen. Ein Blick auf die Differenzfouriersynthese zeigt, daß, wie schon beim ersten Mal, ein Sprung in der Folge der Maxima auffällt und zwar nach Q4. Da gerade noch 4 Atome fehlen, sind Q1 bis Q4 natürlich die heissesten Kandidaten. Es ist an dieser Stelle klug, darauf zu achten, daß die H-Atome an „ihren“ O-Atomen in derselben asymmetrischen Einheit landen. Hierzu dient ein Blick auf die Listeneinträge zu O91 und O92. Tatsächlich zeigt sich bereits bei O91, daß das Restmaximum Q2 ohne Symmetrietransformation im passenden Abstand liegt — zu erkennen an der „0“ vor der „2“, vor der „4“ aber weist die „43“ darauf hin, daß das Q4 der res-Datei nicht unmittelbar an O91 bindet, sondern erst nachdem eine Symmetrieoperation angewendet wurde. Um kein Chaos anzurichten, sollte bei der Editierung von b14.res nicht Q4 in ein H-Atom umgewandelt werden, sondern man kopiert besser aus der lst-Datei das xyz-Tripel, das am Ende der Q-Liste unter „43 4“ tabelliert ist, in die res-Datei und macht dies zum H-Atom. Genauso wird mit O92 verfahren, an das die Restdichten 291 und 403 binden (ab jetzt wird diese Kurzschreibweise verwendet, bei der der Symmetriecode als Hochzahl vor die Laufnummer der Restdichte gesetzt wird). In der fertig editierten res-Datei, abgespeichert als
b14.ins
, sind außer den vier Wasser-H-Atomen noch weitere Anweisungen eingefügt. Insgesamt vier DFIX-Anweisungen (Distance FIX), mit denen Abstände erzwungen werden können, sind eingefügt. Die erste DFIX-Anweisung (DFIX 31 .01 O91 H911 O91 H912) liest sich wie folgt: Erzwinge Atomabstände zu 1 × freie Variable 3 mit einer Standardabweichung von 0.01 für alle nachfolgenden Atompaare, hier also O91 zu H911 und O91 zu H912. Der Startwert für die freie Variable 3 ist 0.83 Å entsprechend dem Mittelwert für röntgenographisch ermittelte O-H-Bindungsabstände. Die folgende DFIX-Anweisung bewirkt dasselbe für das zweite Wassermolekül. Die dritte DFIX-Anweisung fixiert den H-H-Abstand im ersten Wassermolekül auf das 1.57fache der freien Variable 3, wodurch ein Winkel von 105� erzwungen wird. Es spricht nichts dagegen, die DFIX-Anweisungen paarweise zusammenzufassen (DFIX 31 .01 O91 H911 O91 H912 O92 H921 O92 H922 und DFIX 31.57 .02 H911 H912 H921 H922). Die Frage ist nur: Was soll das alles überhaupt?
Röntgenographisch bestimmte Wasserstofflagen. Röntgenographisch bestimmte Atomlagen entsprechen dem Schwerpunkt der Elektronendichte. Diese fällt bei den meisten Elementen weitgehend mit dem Kernort zusammen, bei Wasserstoffatomen ist die Abweichung deutlich, da es keine Rumpfelektronen gibt. H-Lagen werden daher bei der Röntgenstrukturanalyse systematisch falsch bestimmt. H-Atome ganz wegzulassen, ist jedoch auch nicht gerechtfertigt, da sie die Elektronenhülle ihrer Bindungspartner merklich deformieren. Ohne den Beitrag von H-Atomen würde man daher in anisotropen Temperaturparameter noch mehr verstecken als so schon. Man sollte so vorgehen, daß alle H-Atome, deren Position sich aus geometrischen Überlegungen ergibt, mit entsprechenden HFIX- und AFIX-Anweisungen und einem einheitlichen Temperaturparameter einbezogen werden. Wasser-H-Atome werden mit DFIX an ihre O-Atome gebunden, am besten im oben gezeigten Stil (O91, H911 und H912 bilden das Wassermolekül):
DFIX 31 .01 O91 H911 O91 H912
DFIX 31.57 .02 H911 H912
FVAR Skalierungsfaktor .05 .83
O91 ...
H911 ... x1 y1 z1 11 21
H912 ... x2 y2 z2 11 21
Die Bedeutung der einzelnen Anweisungen ist im vorigen Abschnitt zusammengestellt. Es wird dann beobachtet, was bei der Verfeinerung aus der freien Variablen 3 wird. Sie sollte nicht zu weit vom Startwert 0.83 Å abweichen (ca. 0.7–0.95 Å)
Benennung von Atomen. Es fällt vielleicht auf, daß die von HFIX erzeugten Namen von H-Atomen von Hand geändert wurden. An sich ist es nämlich egal, wie Atome benannt werden, solange bei SHELXL der Name 4 Buchstaben nicht überschreitet. Daß Na ein Natriumatom ist, merkt SHELXL nicht am Atomsymbol, sondern an der nachfolgenden SFAC-Nummer. Die R-Werte bleiben daher dieselben, auch wenn statt „Na“ „Otto“ hingeschrieben wird. Allerdings wird der arbeitsgruppeninterne Datenverkehr erleichtert, wenn Konventionen eingehalten werden. Die Mitglieder des AK Klüfers werden daher gebeten, ihre Atome nach einem
einheitlichen Schema
zu benennen.
Wasserstoffbrückenbindungen. Meistens gelingt die Zuordnung von H-Atomen nicht ganz so zwanglos wie bei b14. Eine große Hilfe ist die HTAB-Anweisung, die mit den Wasser-H-Atomen in die ins-Datei eingefügt wurde. Sie führt zu einer Liste aller Wasserstoffbrückenbindungen, die von den H-Atomen an O- oder N-Atomen ausgehen. Wir kommen bei der abschließenden Betrachtung der Struktur darauf zurück. Man beachte auch eine Besonderheit der HFIX-Anweisung. Üblicherweise wird eine Anweisung unverändert von der ins- in die res-Datei übernommen. Anders bei HFIX. Diese Anweisung erzeugt neue Anweisungen und zwar die verschiedenen AFIX-Anweisungen, in denen HFIX in erweiterter Form enthalten ist.
Anpassen von x und y im Wichtungsschema, “goodness of fit”, die OMIT-Anweisung. Zum ersten Mal wurde für die aktuelle ins-Datei die WGHT-Anweisung angepaßt. SHELXL schlägt eine optimierte WGHT-Anweisung anhand statistischer Kriterien vor und schreibt diese neben der alten Anweisung in die res-Datei. Der Ersatz der pauschalen Anweisung WGHT 0.1 durch die Vorgeschlagene empfielt sich am Schluß der Verfeinerung, wenn schon H-Atome und anisotrope Schwingungsparameter berücksichtigt sind. Die letzten Verfeinerungen werden dann einige Male wiederholt, bis die Parameter auf der WGHT-Anweisung konvergiert sind. Nachdem dies geschehen ist (was dem wR2-Wert nicht unbedingt gut tun muß — um diesen glattzubügeln, ist WGHT 0 schwer zu schlagen), sollte ein weiterer Güteparameter, S, der “goodness of fit” (GooF in der lst-Datei) nahe bei 1 liegen. Ein stark von 1 abweichender Wert für S bestraft drei Dinge: (1) ein schlechtes Strukturmodell (das gibt auch schlechte R-Werte), (2) zu wenig Reflexe pro verfeinertem Parameter (gibt prima R-Werte), (3) ein schlechtes Wichtungsschema (umso bessere R-Werte, desto weniger die scheinbare Präzision starker Reflexe korrigiert wird).
Schauen wir nach diesen zahlreichen Ergänzungen zur ins-Datei auf das Ergebnis in
b14.lst
und
b14.res
. Das Wichtungsschema ist noch nicht zur Konvergenz gelangt. Daher wird noch einmal die in der Res-Datei vorgeschlagene WGHT-Anweisung anstelle der alten eingesetzt. Außerdem wurde eine etwas heikle Anweisung eingefügt, die — wenn überhaupt — erst ganz zum Schluß in Betracht gezogen wird. Es handelt sich um OMIT-Anweisungen, durch die einzelne Reflexe aus dem Datensatz eliminiert werden. Da dies eine besonders einfache Methode zur R-Wert-Verbesserung darstellt, sollte sehr umsichtig damit umgegangen werden. Ein nachvollziehbares Vorgehen, um beschädigte Reflexe auszusortieren, besteht darin, in der lst-Datei die Liste der “Most Disagreeable Reflections” anzuschauen und zwar die Spalte “Delta(F^2)/esd”. Ist der Anstieg von unten nach oben nicht einigermaßen linear, sondern weisen die ersten Reflexe deutlich größere Abweichungen auf, sollten diese aus dem Datensatz entfernt werden. Die entsprechende Anweisung lautet OMIT h k l. Mit der so modifizierten Datei
b14.ins
wird erneut verfeinert (
b14.lst
und
b14.res
). Die Änderung ist nun klein und die Strukturanalyse kann abgeschlossen werden. Ein letztes Mal wird die WGHT-Anweisung aktualisiert und es werden drei weitere Anweisungen zugefügt: ACTA legt einen “Crystallographic Information File” (CIF) an, der für die Publikation und Archivierung nützlich ist; BOND und CONF erzeugen Listen mit Atomabständen, Bindungswinkeln und Torsionswinkeln, die sowohl in der lst- als auch in der cif-Datei erscheinen. Zum letzten Mal läuft SHELXL über
b14.ins
und erzeugt
b14.lst
und
b14.res
. Ferner
b14.cif
sowie
b14.fcf
mit einer Gegenüberstellung der Quadrate der beobachteten und berechneten Strukturfaktoren in der CIF-Syntax (wird von manchen Zeitschriften verlangt).
Damit ist Schluß mit der Verfeinerei. Gehen wir anhand der lst-Datei den Stand der Dinge abschließend durch: Alle Atome einschließlich der H-Atome wurden lokalisiert. Das am Anfang unvermeidliche Gemecker “** Cell contents from UNIT instruction and atom list do not agree **” bleibt aus. Die Verfeinerung konvergiert und es werden keine auffälligen Korrelationen (> 0.9) unter “Largest correlation matrix elements” aufgelistet. wR2 ist ganz gut, obwohl bis zu R(int) noch etwas Luft gewesen wäre, S ist mit 0.9 nicht so ganz berauschend (zwischen 0.95 und 1.05 wäre schon ganz ordentlich). Mit 1552 Reflexen wurden 111 freie Parameter verfeinert. In Ordnung ist in einer zentrosymmetrischen Struktur ein 10:1- oder größeres Verhältnis. Auf die Tabelle “Principal mean square atomic displacements U” wird im Abschnitt „ORTEP“ näher eingegangen. Die Tabelle “Bond lengths and angles” pflegt im Gegensatz zu den Abstandstabellen im Fourierteil vollständig zu sein. Eisen ist demnach quadratisch-planar koordiniert, Natrium liegt in einer 5+1-Koordination vor. Die Tabelle der Wasserstoffbrückenbindungen ist sehr erfreulich, indem allen H-Atomen eine Akzeptorfunktion zugeordnet ist. Alle Abstände und Winkel sind normal, wobei die Alkoxid-O-Atome O2 und O3 erwartungsgemäß zu kürzeren Bindungen führen als das Ether-O-Atom O1. Die höchsten und die niedrigsten Restmaxima zeigen, daß keine Atome übersehen oder zuviel gelegt wurden (Letzteres übersteht normalerweise schon nicht die Verfeinerung). Nachdem alles in Ordnung ist, sollte eine Darstellungsweise nicht fehlen, die mit einem Blick erkennen läßt, ob vielleicht doch nicht alles stimmt. Hierzu stellen wir die Schwingungsellipsoide graphisch dar.
Anisotrope Temperaturparameter, ORTEP. Im Laufe der Verfeinerung wurde von isotropen auf anisotrope Temperaturparameter umgestellt. Einem Atom in allgemeiner oder spezieller Lage der Lagesymmetrie 1 werden dabei sechs unabhängige Variable für die Schwingung zugeordnet (drei für die Achslängen eines Ellipsoids, drei für die Richtungen der Hauptachsen). Bei jeder Strukturanalyse ist zu beachten, daß sich in diesen sechs Parametern sehr schön alles Mögliche verstecken läßt, was nichts mit Temperaturschwingung zu tun hat. Stellen Sie sich vor, daß ein Oxolandiolatligand in der Briefumschlagkonformation vorliegt, in der das Ringsauerstoffatom die weggeklappte Ecke ist. In einem ungestörten Kristall wären dann alle O-Atome mit den Lageparametern x, y, z ein einheitlicher Weise weggeklappt — wir nennen diese Richtung „oben“. „Fehlgeordnung“ läge dann vor, wenn statt dessen in statistischer Weise ein Teil der O-Atome nach oben und der andere Teil nach unten geklappt wäre (geschieht es in geordneter Weise, daß aus oben-oben-oben-oben-... oben-unten-oben-unten-... wird, so spricht man von einer „Überstruktur“; bei dieser erscheinen zwischen den starken Röntgenreflexen der „Subzelle“ weitere Reflexe, während sich bei der Fehlordnung lediglich der diffuse Untergrund um die regulären Reflexe erhöht). Das Phänomen Fehlordnung wird in späteren Beispielen näher erläutert. Welche Unregelmäßigkeiten aber zeigen die aniosotropen Temperaturparameter, wenn sie von Effekten außer Schwingung beeinflußt werden? Man sollte beachten, ob nicht (1) alle Ellipsoide unerwartet (a) klein oder (b) groß ausfallen, (2) einzelne Ellipsoide unerwartet klein oder groß ausfallen, (3) alle Ellipsoide in ein und derselben Richtung auffällig stark von der Kugelform abweichen, (4) einzelne Ellipsoide auffällig stark von der Kugelform abweichen.
Im Fall 1a fehlt meist eine Absorptionskorrektur oder die angewandte Absorptionskorrektur ist ungenügend. Beachten Sie, daß für eine zufriedenstellende Korrektur der Besatz der Elemetarzelle bekannt sein muß. Wenn bei der Strukturanalyse etwas anderes herauskommt als zu Beginn angenommen, muß die Absorptionskorrektur wiederholt werden. Fall 1b könnte ähnlich liegen, hier aber können sich weitere Probleme mit streuschwachen Kristallen verbergen. Fall 2 sollte man bereits während der Strukturanalyse im Auge behalten. Ein hoher Temperaturparameter entspricht einer Verringerung der Elektronendichte auf einer Punktlage. Wird eine Lage falsch belegt, wird zum Beispiel anstelle eines Kohlenstoffatoms (6 Elektronen) ein Stickstoffatom (7 Elektronen) gelegt, so wird dessen Temperaturparameter unerwartet hoch ausfallen und umgekehrt. Einzelne „scheibenförmige“ Atome, bei denen eine Ellipsoidhauptachse nahe 0 ist (der noch ungünstigere und physikalisch sinnlose Fall, daß bei der Verfeinerung negative Werte herauskommen, führt zur Fehlermeldung “** NON POSITIVE DEFINITE **” in SHELXL) weisen am Ende einer ansonsten zufriedenstellenden Strukturrechnung auf beschädigte Reflexe hin (siehe oben: OMIT). Fall 3 kann in der graphischen Darstellung, von der gleich die Rede sein wird, sehr lustig aussehen. Wenn alle Atome in der gleichen Richtung zu „Zigarren“ gestreckt sind, meint man seine Struktur bei heftigem Sturm zu sehen. Die Ursache ist eine fehlende oder fehlerhafte Absorptionskorrektur bei stark anisotroper Kristallgestalt (Nadel oder Plättchen). Bei Fall 4 schließlich kommt die erwähnte Fehlordnung in Frage. Eine grobe Fehlordnung in der oben beschriebenen Weise wird sich in langgestreckten Ellipsoiden äußern, die beide Lagen umschließen, über die das betreffende Atom fehlgeordnet ist. Häufig wird SHELXL hier vorschlagen, die mittlere Lage aufzuspalten, wobei xyz-Werte der beiden unterbesetzten Lagen für das eine Atom vorgeschlagen werden (...).
Besonders schnell bemerkt man solche Unregelmäßigkeiten in der graphischen Darstellung. Das klassische Werkzeug hierzu ist ORTEP, für das die Dateneingabe nicht näher beschrieben wird, da inzwischen eine graphische Benutzerschnittstelle zur Verfügung steht, die dem üblichen Standard folgt. Ein
ORTEP-Bild für b14
zeigt, daß man über die Fälle 1a und 4 nachdenken könnte. Fall 1a ist schwach angedeutet; die Temperaturparameter sind an der unteren Grenze, aber es könnte in Ordnung gehen. Man würde also die Absorptionskorrektur zur Sicherheit überprüfen. [[...]] Bei der graphischen Darstellung würde man der Übersichtlichkeit halber größere Atome zeichnen. Hierzu wird der ORTEP-Parameter „Aufenthaltswahrscheinlichkeit“, der standardmäßig auf 50 % eingestellt ist, zum Beispiel auf 80 % erhöht. Die Schwingungsellipsoide beschreiben dann den Raum, in dem sich der Atomschwerpunkt mit einer Wahrscheinlichkeit von 80 % aufhält. Die Möglichkeit, diesen Parameter zu manipulieren, bedeutet natürlich auch, ihn zu dokumentieren. Wenn Sie im Bericht über eine Strukturanalyse ein ORTEP-Bild zeigen, gehört die Aufenthaltswahrscheinlicheit in die Bildunterschrift. Um die Struktur abzubilden, sollte man natürlich eine signifikante Baueinheit zeigen. In diesem Beispiel ging es um die
Struktur des komplexen Anions
, das am Besten mit seiner 1-Symmetrie dargestellt wird. Im Bild wurden außerdem Atomsymbole angebracht und zwar „von Hand“, da die automatische Zuweisung von Symbolen nicht so toll aussieht (hierfür gibt es mehrere Möglichkeiten: ORTEP für Windows bietet eine solche Routine an; hier wurde das ORTEP-Bild ohne Symbole als Postscript-Datei nach CorelDraw überführt und dort der Rest erledigt). Auch wenn es bei der Publikation üblich ist, Bilder zu erzeugen, in denen ein Teil der Struktur fehlt (hier Natrium und Wasser), sollte stets zur Kontrolle der Strukturanalyse auch das vollständige ORTEP-Bild betrachtet werden.
Das ORTEP-Bild von b14 ist weitgehend normal. Es treten deutliche Abweichungen von der Kugelgestalt der Atome auf, aber fast immer ist es plausibel, in welcher Richtung größere Temperaturbewegung erfolgt. Ein Blick auf die Atome des Chelatfünfrings mit Eisen zeigt, daß diese jeweils in der Richtung stärker schwingen können, bei denen die Länge der Bindungen am wenigsten verändert werden — so, wie es ein Modell vorhersagen würde, in dem nach Art der Schwingungsspektroskopie Bindungen als Federn betrachtet werden. In einem solchen Modell wäre die Auslenkung senkrecht zu den Bindungen am leichtesten möglich, und genau dies zeigen die Schwingungsellipsoide auch an. Lediglich C2 sieht nicht ganz gesund aus. Die ausladenste Schwingung zeigt das Ring-Sauerstoffatom O1, aber auch hier ist die größte Auslenkung senkrecht zu den Bindungen. In einer späteren Übung wird gezeigt, wie eine ausladende Schwingung von einer Fehlordnung unterschieden werden kann.
Übersicht über alle Dateien.
Um den Gang der Strukturanalyse noch einmal schrittweise nachzuvollziehen, verkleinern Sie am Besten das Browser-Fenster so weit, daß Sie nur noch diese Tabelle sehen, machen das Hilfsfenster so groß wie es geht, und klicken sich dann schrittweise durch die Dateien.
Weitere Dateien:
b14.hkl,
SCHAKAL-Bild nach 1,
SCHAKAL-Bild mit Atomnamen,
ORTEP nach 7 (50 % Aufenthaltswahrscheinlichkeit),
ORTEP nach 7 (80 % Aufenthaltswahrscheinlichkeit),
b14.cif,
b14.fcf.
Beispiel 2: [(en)2Pd2(C4H6O4)] · 10 H2O in der Raumgruppe P 1
In diesem Beispiel lernen Sie eine Struktur mit Vektormethoden zu lösen. Mit Hilfe von SHELXS wird eine Pattersonsynthese berechnet, aus der die Schweratomlage bestimmt wird. Weiter geht es dann mit Differenzfouriersynthesen.
Pattersonsynthese. Das Beispiel ist wieder eine Strukturbestimmung, bei der es keine Probleme gibt. Die Elementarzelle ist nun mit zwei Schweratomen gefüllt. Da auch jetzt keine chiralen Edukte eingesetzt wurden, liegt wieder die Raumgruppe P 1 nahe. Wir entnehmen einer Pattersonsynthese, daß nicht etwa zwei spezielle Punktlagen mit Schweratomen belegt sind, sondern daß die beiden Atome in allgemeiner Lage liegen. Aus derselben Pattersonsynthese errechnen wir die freien Lageparameter x, y und z. In der Praxis würde man ähnlich vorgehen, da auch bei diesem Beispiel Direkte Methoden nicht mit den Standardeinstellungen zum Ziel führen. Bevor sich der Eindruck festsetzt, die vielgelobten Direkten Methoden taugten nicht viel: Sie setzen eine möglichst gleichmäßige Verteilung der Elektronendichte in der Elementarzelle voraus — genau das aber ist bei einfachen Lehrbeispielen nicht gegeben, bei denen ein oder zwei Schweratome in der Elementarzelle das Beugungsbild dominieren.
Der Kristall. Die untersuchte Substanz ist [(en)2Pd2(C4H6O4)] · 10 H2O und wurde von G. Kettenbach in Form gelber Kristalle aus Lösungen von Erythrit (C4H10O4, meso-Butan-1,2,3,4-tetraol) im Celluloselösungsmittel „Pd-en“, einer wäßrigen Lösung von [(en)Pd(OH)2], isoliert. Geplant war die Synthese eines Zweikernkomplexes, bei dem zwei (en)Pd-Einheiten an das Tetraol im Sinne eines Bis-diols koordinieren. Die Elementaranalyse zeigt, daß pro mol vollständig deprotoniertem Erythrit 2 mol Palladium, 2 mol Ethylendiamin und 10 mol Wasser enthalten ist. Die Summenformel wäre dann C8H44N4O14Pd2. Die Dichte der Kristalle wurde nicht bestimmt.
Die Metrik. Die Abmessungen der Elementarzelle werden vom Diffraktometer wie folgt ausgegeben:
a = 7.3343(15), b = 9.0475(15), c = 9.9891(18) Å, α = 82.812(15), β = 78.375(17), γ = 68.891(15)�
Zahl der Formeleinheiten. Das Volumen der Elementarzelle beträgt gerundet 605 Å3. Dividiert durch 19 ergeben sich 31.8 Atome pro Elementarzelle. Aus der Summenformel ergeben sich 28 Nichtwasserstoffatome. Z ist also auch in diesem Beispiel 1, in der Elementarzelle finden wir diesmal also zwei Schweratome.
Wir legen die Schweratomlage fest. Ganz so einfach wie beim ersten Beispiel ist es diesmal nicht. Bleiben wir aus denselben Gründen wie vorher zuerst einmal bei der zentrosymmetrischen Raumgruppe P 1 (beachten Sie die trikline Metrik!), dann würden wir bei einem trial-and-error-Anlauf zwei spezielle Punktlagen mit je einem Metallatom belegen — aber das wäre schon zuviel der Probiererei. Wahrscheinlicher ist, daß die beiden Palladiumatome die zweizählige allgemeine Lage besetzen, so daß wir vor der Frage stehen, wie man an Startwerte für die drei freien Variablen x, y und z kommen kann. Hier darf man von Kleinste-Fehler-Quadrate-Rechnungen nicht zuviel erwarten. Einfach irgendwo ein Atom hinzulegen und loszuverfeinern ist keine gute Idee. Stattdessen versuchen wir es mit ...
Vektormethoden. Die Fouriertransformation der F-Werte einschließlich der korrekten Vorzeichen führt zur Elektronendichteverteilung. Gemessen wurden aber nur die F2-Werte. Was erhält man eigentlich, wenn man in stiller Verzweiflung eine Fouriertransformation über F2 rechnet? — Eine Vektorkarte. Die Elementarzelle ist nun mit Maxima gefüllt, deren Koordinaten den Spitzen interatomarer Vektoren entsprechen. Am besten macht man sich das an einem Beispiel klar. Schauen Sie hierzu eine
Zeichnung
an. Dargestellt sind vier Elementarzellen in der Raumgruppe P 1. Die Projektionsrichtung ist [001] (c weist also nach hinten). Der Winkel γ soll zufällig 90� sein, α und β nicht, daher liegen a und b nicht in der Zeichenebene, sondern nur ihre Projektionen aP und bP. Die Bezeichnung des Ursprungs einer Elementarzelle ist in den Internationalen Tabellen leider nicht einheitlich; meist findet man „0“, manchmal aber auch „o“ (von origo, lat.: Ursprung); im Beispiel wurde „o“ gewählt. Wir betrachten nun die rechte obere Zelle, die dadurch hervorgehoben ist, daß die Symmetriezentren als kleine offene Kreise eingezeichnet sind (vergleichen Sie mit dem Bild in den Internationalen Tabellen). Die Zelle enthalte ein Atom mit den Koordinaten x, y, z. Durch Punktspiegelung am Symmetriezentrum in 0, 0, 0 entsteht ein Bildobjekt mit den Koordinaten –x, –y –z, das in der linken unteren Zelle liegt. Das gesamte Muster ergibt sich nun, indem die Elementarzelltranslation auf das Originalobjekt (im einzelnen: [100], [010] und [110]) und auf das Bildobjekt ([100], [010] und [110]) angewendet werden. In der oberen rechten Zelle befinden sich dann zwei Objekte, eines bei x, y, z und eines bei 1 – x, 1 – y und 1 – z, wobei die Lage des letzteren auch direkt durch Punktspiegelung an 1 in ½, ½, ½ erhalten werden kann.
Zu einer Vektorkarte gelang man nun, indem alle Vektoren zwischen der Lage x, y, z und den umliegenden Lagen eingezeichnet werden. Im Bild sind die Vektoren durch Doppelpfeile dargestellt, die jeweils den Vektor Atom 1 → Atom 2 und den Vektor Atom 2 → Atom 1 zusammenfassen. Werden Vektoren weggelassen, die der einfachen Translation um einen Basisvektor der Elementarzelle entsprechen, so ergeben sich die vier eingezeichneten Vektorpaare. Im nächsten Schritt stellen wir die interatomaren Vektoren in derselben Elementarzelle wie die Struktur dar. Hierzu betrachten wir zuerst den Vektor, der von der Lage –x, –y, –z zur Lage x, y, z verläuft. Sein Betrag entspricht der Differenz der Koordinaten, also 2x, 2y, 2z. In der Zeichnung ist dieser Differenzvektor rot eingezeichnet. Dasselbe wiederholt man nun mit allen anderen interatomaren Vektoren. Das Ergebnis macht man sich am Besten graphisch klar: Jeder rot eingezeichnet Vektor wird mit seinem Fußpunkt auf eine der vier Zellecken gelegt und zwar so, daß seine Spitze in der rechten oberen Zelle zu liegen kommt. Ist dies mit allen vier Vektorpaaren geschehen, so haben sich nur zwei Lagen für die Vektorspitzen ergeben, und zwar die schon genannte Lage 2x, 2y, 2z und die Lage 1 – 2x, 1 – 2y, 1 – 2z, die durch das Symmetriezentrum bei ½, ½, ½ erzeugt ist. Es wird nun der Nutzen einer F2-Fouriersynthese, einer „Patterson-Synthese“, klar: Gelingt es, aus der mit den roten Patterson-Maxima gefüllten Elementarzelle das erzeugende, schwarze Atomlagenmuster zu rekonstruieren, ist die Struktur gelöst. Bei einer größeren Zahl von Atomen in der Zelle scheint dies eine unlösbare Aufgabe zu sein, aber: die Höhe eines Pattersonmaximums ist dem Produkt der Elektronenzahlen der beiden Atome proportional, die den Vektor bilden. Die Pattersonsynthese ist also bestens geeignet, die Schweratomlagen zu bestimmen. Nun zur Praxis: Die Datei
pd1.ins
enthält die PATT-Anweisung, die zur Berechnung einer Pattersonsynthese führt. In
pd1.lst
ist das Ergebnis abzulesen. Schauen Sie dazu auf die mit X Y Z Weight Peak Sigma Length beginnende Tabelle, in der die interatomaren Vektoren nach fallendem Betrag („Peak“) aufgelistet sind. Das höchste Maximum, auf das alle folgenden Maxima normiert sind, ist das mit den Koordinaten 0, 0, 0. In diesem Maximum sind alle Vektoren zusammengefaßt, die durch eine Zelltranslation zustande kommen (vulgo: die Vektoren aller Atome zu sich selbst). Für das nächste Maximum (Peak: 456) ist das Koordinatentripel 0.3764 0.2023 0.2188 angegeben. Beachten Sie, daß das dann folgende Maximum von deutlich kleinerem Betrag ist (Peak: 152). Das 456er Maximum sollte einem Pd-Pd-Vektor entsprechen, die folgenden Maxima sollten Vektoren zwischen Pd und leichteren Atomen entsprechen. Dementsprechend sind die Vektoren 3 und 4 mit ca 2 Å (Spalte „Length“) so lang, wie es für eine Pd-N- oder Pd-O-Bindung erwartet werden darf. Für die Strukturlösung wichtiger ist der Pd-Pd-Vektor. Da dieser symmetrieerzeugt ist, läßt sich unmittelbar eine Pd-Lage ableiten. Division der Vektor-Koordinaten durch 2 führt nämlich (in jeder zentrosymmetrischen Raumgruppe!) vom Vektor 2x, 2y, 2z zur erzeugenden Atomlage x, y, z — und schon haben wir eine Startbesetzung der Elementarzelle, um in die Differenzfouriersynthese einzusteigen. Bevor es weitergeht, überlegen Sie zur Übung zwei Dinge: (1) wie hätte die Pattersonsynthese ausgesehen, wenn nicht die allgemeine Punktlage mit 2 Pd-Atomen belegt wäre, sondern zwei spezielle Punktlagen mit jeweils einem, zum Beispiel 1 Pd in 1 a 0, 0, 0 und 1 Pd in 1 h ½, ½, ½; (2) die Elementarzelle enthält zwei Pattersonmaxima; hätten wir auch das andere Maximum wählen können, um nach Division der Koordinaten durch 2 die Pd-Lage zu ermitteln?
Automatische Interpretation der Pattersonsynthese. Es gibt Algorithmen, die den Schritt von der Vektorkarte zum erzeugenden Muster zu tun. SHELXS bedient sich solcher Verfahren, um einen Strukturvorschlag zu machen. Allerdings kommt bei diesem Beispiel nichts Gescheites heraus. Hier wäre nur die Erfahrung zu wiederholen, daß Strukturen mit nur einem Schweratom — vor allem im triklinen Kristallsystem — für automatisierte Lösungsverfahren keine Freude sind.
Leichtatomlagen. Es geht wie beim ersten Beispiel mit einer Differenzfouriersynthese weiter, um die Lage der leichteren Atome zu bestimmen. Beachten Sie beim Betrachten der
ins-Datei
zwei Einzelheiten: (1) Den Besetzungsfaktor k für die Pd-Lage. Pd belegt die allgemeine Lage, k ist daher 1. Der Wert soll bei der Verfeinerung nicht verändert werden, weswegen 10 addiert werden, was zur Angabe 11 auf der Pd-Atomanweisung führt. (2) Im Gegensatz zu Beispiel 1 wird eine L.S.-Anweisung eingefügt. Da Pd allgemein liegt, gibt es jetzt mit x, y und z nämlich etwas zu verfeinern. Die
lst-Datei
zeigt das Ergebnis. Bereits nach der Lokalisation eines einzigen Atoms beträgt R1 ca. 0.26. Vergleichen Sie mit dem deutlich schlechteren R-Wert in Beispiel 1. Dadurch daß Pd allgemein liegt und ein 4d-Element mit entsprechend hoher Ordnungszahl ist, ist der Anteil des Palladiums an der gesamten Elektronenzahl deutlich höher als zuvor bei Eisen. Die Differenzfouriersynthese zeigt wie im ersten Beispiel einen deutlichen Abbruch, der in der
res-Datei
besonders auffällt. Er liegt hier nach der Restdichte Q13; es sieht also wieder gut aus: in der Elementarzelle werden 28 Atome erwartet, in der asymmetrischen Einheit also 14. Sowohl die Pd-Lage als auch die Lage von Q1 bis Q13 sind allgemein, so daß insgesamt die gesuchten 14 Atome zusammenkommen. Schauen wir also wieder auf das
SCHAKAL-Bild
(Q in F umbenannt!). Q3, Q6–8 und Q11 liegen irgendwie so herum, besonders eigenartig aber Rest: Palladium scheint zwar wie üblich quadratisch-planar koordiniert zu sein, allerdings von zwei Molekülen Ethylendiamin anstatt von einem. Bevor wir voreilige Schlüsse ziehen, noch ein Blick auf die Lage der Restmaxima in der Elementarzelle. Hierzu wird g u (generate unit) eingegeben. Im
ergänzten Bild
sieht man nun die Basisvektoren b (rot) und c (grün), während entlang der Projektionsrichtung a an jedem Atom dessen Achsabschnitt entlang a, multipliziert mit 100, angegeben ist (zum Beispiel ist bei Palladium „18“ angegeben entsprechend x ≈ 0.18). Überträgt man nun in Gedanken die Symmetriezentren der Raumgruppe P 1 in das Bild (jede Ecke, jede Kantenmitte, jede Flächenmitte, die Raummitte), so wird deutlich, daß das unterste Atom in einem Chelatfünfring nahe bei dem Symmetriezentrum in 0, 0, ½ liegt. SCHAKAL wird daraufhin angewiesen, die bisher gezeigten Atome durch eine gleiche Zahl zu ergänzen, die durch Punktspiegelung an 0, 0, ½ entstehen. Hierzu lädt man mit u i + Eingabetaste die aktuelle res-Datei neu ein und gibt auf die Frage nach weiteren Anweisungen ein: du -x, -y, 1-z [duplicate] und außerdem wegen der Basisvektoren der Zelle: g u. SCHAKAL zeigt nun ein
vollständiges Molekül
. (Machen Sie sich am Bild klar, warum die Koordinaten der hinzugekommenen Hälfte –x, –y, 1 – z sind.) Es wird deutlich, daß es sich bei den Liganden von Palladium nicht um zwei Ethylendiaminmoleküle handelt, sondern um einen en- und einen Erythrit-Liganden. Das Symmetriezentrum des Erythrits fällt mit einem Symmetriezentrum der Raumgruppe P 1 zusammen, so daß nur 2 C- und 2 O-Atomlagen bestimmt werden müssen, um die vollständige C4O4-Kette festzulegen. Während es nun leicht ist, die Ligandatome um Palladium zuzuordnen, sollte wegen der Restmaxima Q3, Q6–8 und Q11 noch ein Blick in die lst-Datei geworfen werden. Alle diese Maxima weisen Abstände von ca. 2.7 Å zu O-Atomen und untereinander oder etwas größere Abstände zu N-Atomen auf, wodurch es naheliegt, sie als Wasser-O-Atome zu behandeln. Das Ergebnis ist die nächste
ins-Datei.
Da damit zu rechnen ist, daß alle Nichtwasserstoffatome gefunden wurden, wurde gleich eine ANIS-Anweisung mit eingefügt sowie eine HFIX-Anweisung für die C- und N-Atome. Die
lst-Datei
mit dem Ergebnis der Rechnung macht den Eindruck, als ließen sich noch etliche H-Atome der Wassermoleküle lokalisieren. Leider kommt man bei solchen Bemühungen nicht richtig zum Ziel, weshalb das Beispiel 2 an dieser Stelle enden soll. Die Ursache für den Ärger ist übrigens kein seltener. H-Atome lassen sich oft zuordnen, wenn klar ist, was der Donor und was der Akzeptor ist (so auch bei Beispiel 2; in der Umgebung der Akzeptoren O1 und O2 findet man im erwarteten Abstand von ca. 1.8 Å passende Restmaxima). Wird der Kristall dagegen von H-verbrückten Wassermolekülen durchzogen, so tritt die Situation auf, daß die H-Brücken zwischen z. B. zwei Atomen O91 und O92 so aussehen könnten: O91–H···O–H···O–H···O92–H, genausogut, das heißt mit gleichem Energieinhalt aber auch: O91···H–O···H–O···H–O92. Gibt es im Kristall nun nebeneinander Domänen, die das eine oder das andere der beiden Muster realisieren, so „sieht“ der Röntgenstrahl eine gemittelte Anordnung, nämlich: h–O91–h··h–O–h··h–O–h··h–O92–h (h = ½ H) — und genau so etwas scheint im Beispiel 2 vorzuliegen (das prominenteste Beispiel für diesen Fall ist sicher Eis-1h, das normale Eis). Beachten Sie, daß diese wie auch jede andere Kristallpathologie eine klare chemische Ursache hat, hier die Ununterscheidbarkeit von Donor und Akzeptor, der in beiden Fällen Wasser ist.
Zum Schluß bleibt nur noch, die Wichtung anzupassen (
ins-,
lst-
und
res-Datei),
was wiederholt wird (
ins-,
lst-,
res-Datei),
wobei im letzten Durchgang durch die ACTA-Anweisung ein
Crystallographic Information File
und eine
Liste der Strukturfaktoren
angelegt wird. Man beachte die BOND- (Abstände und Winkel), HTAB- (H-Brücken) und CONF-Anweisung (Torsionswinkel) in der letzten ins-Datei. Zur Kontrolle auch hier wieder ein Blick auf die
Schwingungsellipsoide
(60 % Aufenthaltwahrscheinlichkeit); es zeigt sich auch hier, daß die Wasser-Teilstruktur etwas leidet.
Übersicht über alle Dateien.
Um den Gang der Strukturanalyse noch einmal schrittweise nachzuvollziehen, verkleinern Sie am Besten das Browser-Fenster so weit, daß Sie nur noch diese Tabelle sehen, machen das Hilfsfenster so groß wie es geht, und klicken sich dann schrittweise durch die Dateien.
Weitere Dateien:
Prinzip einer Vektorkarte,
SCHAKAL-Bild,
dto. mit Zelle,
dto. mit 1,
CIF,
Strukturfaktorliste
60-%-ORTEP
Beispiel 6: Li[Si(C4H6O3)2(OH)] · H2O in der Raumgruppe P 21/c
In diesem Beispiel wird der Ärger gezeigt, dem man bei einem Datensatz ausgesetzt ist, der von einem Zwillingskristall gewonnen wurde. Der Zwilling in diesem Beispiel gehört zur Sorte „pseudomeroedrisch“. Es wird ferner auf die missym-Routine von PLATON eingegangen.
Es fängt scheinbar normal an: Die Metrik deutet auf das orthorhombische Kristallsystem hin, die systematischen Auslöschungen führen eindeutig zur azentrischen Raumgruppe P m c 21 (No. 26). Die Einordnung ins orthorhombische System wird durch den internen R-Wert bestätigt (9101 hkl gemittelt zu 1565 unique hkl, Ri = 0.052). Direkte Methoden führen jedoch nicht überzeugend zu einer Strukturlösung. Man sieht als Ergebnis nur einen Teil der Struktur (das allein heißt noch gar nichts), aber der CFOM-Wert ist größer als 0.3 (was ernster ist). Neben dem CFOM-Wert passt jedoch noch etwas nicht: Die mittleren Beträge von E2 − 1 (Mean Abs(E*E-1)) entsprechen nicht der Erwartung. Für zentrosymmetrische Daten ist der Erwartungswert 0.968, für azentrische 0.736. Die azentrische Raumgruppe P m c 21 hat in [100] und [010] azentrische und in [001] eine zentrosymmetrische Projektion, für die tabellierte Abfolge 0kl h0l hk0 Rest wäre also zu erwarten: 0.736 0.736 0.968 0.736, es errechnet sich jedoch 1.058 0.897 0.878 0.873, was alles viel zu nahe an der Zentrosymmetrie ist. Da die Struktur keine besonders schweren Atome enthält (ab ca. zweiter Übergangsreihe), ist das bedenklich. Versuche weiter zu rechnen, führen zu nichts Gescheitem, außerdem sind die R-Werte immer recht hoch (> 0.5). Auch wenn der gute innere R-Wert es nicht nahelegt, würde man als Nächstes versuchen, in einer niedersymmetrischen Raumgruppe die Struktur zu lösen (was will man sonst machen?). Hierzu geeignet sind zuerst einmal die Untergruppen von P m c 21, nämlich P 1 1 21 (P 21), P 1 c 1 (P c) und P m 1 1 (P m). Es zeigt sich, dass es in P c schnell vorangeht. Man ist also im monoklinen Kristallsystem gelandet. Da bis hier noch nicht viel Arbeit investiert worden ist, fängt man am Besten noch einmal mit AUSL von vorne an. Das Ergebnis ist wenig originell: P 21/c. Der Rest ist zuerst einmal Routine, die mit
b92.ins
angeworfen wird. Die von den Direkten Methoden
(b92.lst,
b92.res)
vorgeschlagene Si-Lage plus die nächsten 18 Restmaxima zeigen die komplette Struktur, so dass schon beim nächsten Lauf eine anisotrope Verfeinerung sowie an Kohlenstoff gebundene Wasserstoffatome berücksichtigt werden können
(b92.ins,
b92.lst,
b92.res).
Der ORTEP-Plot sieht auch gut aus – aber wR2 hängt bei unglaublichen 0.445! R-Werte geben Auskunft über die Übereinstimmung zwischen Modell und Daten. Ein hoher R-Wert zeigt in der Regel an, dass das Modell nichts taugt. Sieht wie in unserem Fall die Strukturlösung aber chemisch sinnvoll aus, und – vor allem – sieht die anisotrope Verfeinerung so unproblematisch aus, sollte darüber nachgedacht werden, ob nicht die Daten beschädigt sind.
Befassen wir uns zuerst mit dem Modell. Dessen Entstehung war schon etwas seltsam. Alles sprach für das orthorhombische Kristallsystem, unser Modell ist jedoch mit monokliner Symmetrie aufgestellt. Der Fall, dass zu wenig Symmetrie berücksichtigt wurde, lässt sich sehr schnell überprüfen. Das Programm PLATON gibt die Möglichkeit, durch den Befehl „missym“ übersehene Symmetrie zu ermitteln. Sie sollten diese Routine nach jeder Strukturbestimmung laufen lassen! In unserem Fall wird das Programm fündig. Es wird nicht die Raumgruppe P 21/c = P 1 21/c 1, sondern P b c m = P 2/b 21/c 21/m (No. 57) vorgeschlagen, die eine minimale Obergruppe von P 21/c ist („minimal“ ist eine Obergruppe, wenn sie in 1 Schritt, also durch das Hinzufügen möglichst weniger Symmetrieelemente erreicht wird; die Untergruppe ist in diesem Fall eine maximale Untergruppe; beachten Sie, dass in den Internationalen Tabellen bei jeder Raumgruppe deren minimale/maximale Ober-/Untergruppen angegeben sind). Jetzt scheint nichts mehr zu stimmen. Auf die Raumgruppe Pbcm hätten wir ganz zu Beginn stoßen müssen, als wir abgefragt haben, ob Reflexe der Zone 0kl nur für gerade k auftreten (dies ist die Bedingung für eine b-Gleitspiegelebene senkrecht zu [100]). Dies war aber nicht der Fall! Am besten schauen wir die
Elementarzelle
einmal genau an. Eines der Symmetrieelemente, die PLATON entdeckt hat, ist eingezeichnet, nämlich die Spiegelebene senkrecht zu [001]. Man sieht, dass die Ebene nur näherungsweise erfüllt ist. Die Toleranzen sind in PLATON (sinnvollerweise) so weit gesetzt, dass lieber einmal zu oft als einmal zu wenig gewarnt wird. Oder können wir in der Obergruppe viel bessere R-Werte erwarten? Natürlich nicht! Die Strukturverfeinerung in einer Untergruppe tut den R-Werten schließlich eher gut, da ja mehr freie Parameter zur Verfügung stehen. So ganz nebenbei sind wir aber dem Problem des Datensatzes nähergekommen. Auch wenn die Struktur in Pbcm nicht genau zu beschreiben ist, so kommt Pbcm der wahren Symmetrie sehr nahe. In diesem Fall besteht immer der Verdacht, dass der Datensatz durch Zwillingsbildung beeinträchtigt ist. Und das geht so: Bei der spontanen Keimbildung werden die ersten Kristallkeime nach der Oswaldschen Stufenregel in einer energiereichen Hochtemperaturform anfallen – wenn es eine solche Form gibt. Lässt sich eine niedersymmetrische Struktur durch geringfügige Verschiebungen der Kristallbausteine in eine höhersymmetrische Form überführen – so wie hier –, dann ist die höhersymmetrische Form ein sehr guter Kandidat für die Struktur der Hochtemperaturphase. Beim weiteren Keimwachstum kommt es dann zur Umwandlung in die stabile Phase. In unserem Beispiel gehen durch kleine Verschiebungen die Spiegelebenen verloren. Die Wahrscheinlichkeit, dass dies in allen Volumenabschnitten des Kristalls gleichsinnig erfolgt, ist nicht sehr hoch. Man kann sich vielmehr leicht vorstellen, dass es Domänen der einen Verzerrungsvariante neben solchen mit spiegelbildlicher Verzerrung gibt – entsprechend der 50:50-Wahrscheinlichkeit, dass ein ungeschickter Reiter nach links oder nach rechts von seinem (spiegelsymmetrischen) Pferd fällt. Wir können also als Fazit damit rechnen, dass unsere Substanz nicht in Form von Einkristallen vorliegt, sondern dass sich „Zwillinge“ gebildet haben – dummerweise aber nicht in Form irgendwelcher schiefer Verwachsungen, die man sofort leicht erkennt und vor der Messung mit dem Skalpell trennen kann. Erinnern Sie sich vielmehr daran, dass – als Folge der großen Nähe zur höheren Symmetrie – der monokline Winkel β sehr nahe bei 90� lag. Die Spiegelung senkrecht zu [001] bildet daher das Gitter – und damit auch das reziproke Gitter – auf sich selbst ab. Sie merken also zuerst einmal ihrem Beugungsmuster überhaupt nichts an, aber alle Reflexintensitäten sind durch Überlagerung verfälscht! Bevor wir versuchen, dies zu beheben, sollten wir uns klar machen, wie sich im allgemeinen Fall abschätzen lässt, zu welcher Störung die Nähe einer Struktur zu einer höhersymmetrischen Variante in einer höhersymmetrischen Raumgruppe führt. Es gilt die Regel, dass die Natur dazu neigt, die verlorenen Punktsymmtrieelemente in makroskopischer Zwillingssymmetrie zu konservieren. Wir achten also auf verschwindende Punktsymmetrieelemente: Beim Übergang P 2/b 21/c 21/m → P 1 21/c 1 verschwinden die 2-zählige Achse entlang [100] und die schon genannte Spiegelebene mit der Normalen [001]. Wir dürfen davon ausgehen, dass auch die 2-zählige Achse zu einem ähnlichen Phänomen führen wird. Für die Praxis der Korrektur ist dies in der Regel egal. Die folgende Rechnung führt unter Annahme eines „Achsenzwillings“ zum gleichen Ergebnis wie im Fall des hier angenommenen „Ebenenzwillings“. Man macht folgendes: In SHELXL wird eine TWIN-Anweisung eingefügt, auf der zeilenweise die Transformationsmatrix angegeben ist, welche die ursprünglichen Basisvektoren der Elementarzelle in das Vektortripel der Zwillingszelle überführt. Spiegelung an (001) bildet [100] und [010] auf sich selbst ab, lediglich [001] wird in [001] überführt. Die TWIN-Anweisung lautet:
TWIN 1 0 0 0 1 0 0 0 -1
BASF .5
Als zweite Anweisung folgt eine batch-scale-factor-Anweisung, die einen Startwert für den zu verfeinernden Volumenanteil des Zwillingsanteils vorgibt. Wird die BASF-Anweisung weggelassen, so wird von einem konstanten 1:1-Verhältnis der Anteile ausgegangen. Mit diesen beiden Anweisungen verschwinden bei unserer Siliciumverbindung alle Probleme
(b92.ins,
b92.lst, | b92.res).
Um die Strukturbestimmung zu beenden, sind nur noch die an O-gebundenen Wasserstoffatome zu lokalisieren (es sind die drei stärksten Restmaxima) und die Wichtung ist anzupassen. Das Ergebnis ist im
CIF
zusammengefasst. Die erste Einschätzung, dass die Struktur in Ordnung ist, aber nicht der Datensatz, wird sehr schön deutlich, wenn die ORTEP-Plots der fertigen Strukturbestimmung und der Daten mit R > 0.4 verglichen werden:
ORTEP-Plot ohne (oben) und mit (unten) TWIN. Zwischen den beiden Bildern liegen ca. 0.4 in wR2!
Zum Schluss noch eine Übung zur Zwillingsbildung: Stellen Sie sich vor, eine Struktur ist in der Raumgruppe P 1 zu beschreiben. Beim Kristallwachstum wurde die Symmetrie 1 zerstört, so dass Zwillinge gebildet werden, die zu gleichen Teilen aus der gegebenen Struktur und ihrem durch Inversion entstandenen Bild bestehen (man spricht dann von einem „Inversionszwilling“). In welcher Weise wird die Strukturanalyse beeinträchtigt sein?
Übersicht über alle Dateien.
Um den Gang der Strukturanalyse noch einmal schrittweise nachzuvollziehen, verkleinern Sie am Besten das Browser-Fenster so weit, daß Sie nur noch diese Tabelle sehen, machen das Hilfsfenster so groß wie es geht, und klicken sich dann schrittweise durch die Dateien.
Weitere Dateien:
b92.hkl, ausl.lis, Elementarzelle, ORTEP-Plot ohne (oben) und mit (unten) TWIN.
Allgemeiner Teil
Kristallzucht
3 Beispiele mit Bildern, wie man bei uns so etwas macht.
Kristallauswahl
Polarisationsmikroskopie: Doppelbrechung, optische Achsen in Durchstrahlungsrichtung, Zwillingsgrenzen, kubische Kristalle
Optimale Größe, Schneiden vs. freigewachsende Kristalle, Folgen mechanischer Belastung
Erste Röntgenaufnahmen: Auflösung (Beugungsbilder guter und mieser Kristalle zeigen)
Unsere Röntgengeräte
Vor- und Nachteile, Kühlung, Phasenumwandlungen beim Abkühlen, statische Fehlordnung beim Abkühlen, Auswahl der Strahlung (Mo vs. Cu; Drehanode vs. geschlossene Anode
Bestimmung der Metrik, Vermuten des Kristallsystems
Korrelation Metrik–Kristallsystem
Bestimmung der Laueklasse
Systematische Auslöschungen, Auslöschungssymbol
Raumgruppenbestimmung
Einschränkungen bei chiralen Substanzen
E2–1-Statistik (alle hkl, einzelne Zonen)
Strukturlösung mit Direkten Methoden
Differenz-Fourier-Synthese
Phasenbestimmung durch 1 Schweratom
Verfeinerung, Temperaturparameter falsch zugeordneter Atome
Absolute Struktur
Zelltransformationen
Besonders im orthorhombischen Kristallsystem kommt es oft vor, dass eine Transformation einer Struktur von einer Nicht-Standardaufstellung in eine Standardaufstellung nötig ist oder umgekehrt. Es wird an einem Beispiel gezeigt, wie vorzugehen ist. Das Beispiel verwendet keine vollständigen Daten, sondern nur einen Ausschnitt, da sich die Einzelschritte dann besser zusammenfassen lassen. Die Ausgangs-Raumgruppe ergibt sich bei orthorhombischer Laue-Symmetrie aus der systematischen Auslöschung 0kl nur für l = 2n. Hierzu gehört laut International Tables, S. 43, die Raumgruppe Pc2m (28), die nicht fettgedruckt ist, das heißt, es handelt sich nicht um die Standardaufstellung der Raumgruppe mit der Nummer 28. Schlägt man im Hauptteil der Tabellen unter Nr. 28 nach oder schaut gleich auf den Seiten 56–58 die möglichen Aufstellungen nach, so ergibt sich Pma2 als Standardaufstellung. Es spart in aller Regel Arbeit, an dieser Stelle den Datensatz zu transformieren. Man geht mit Hilfe der Tabellen auf S.56–58 folgendermaßen vor: (1) Unter der Nummer 28 sucht man die aktuelle Aufstellung Pc2m. (2) Der Tabellenkopf zeigt, das bei dieser Aufstellung die Basisvektoren der Standardaufstellung abc in der Reihenfolge bca vorliegen. (3) Das bedeutet, dass der Standardbasisvektor a in der aktuellen Zelle an dritter Position liegt, der Standardbasisvektor b an der ersten und c an der zweiten Position. Um die Gitterkonstanten und die hkl-Werte von der aktuellen in die Standardaufstellung zu transformieren, ist also (4) die aktuelle Position 3 (001) zur ersten zu machen, die aktuelle erste (100) zur zweiten, und die aktuelle zweite (010) zur dritten. Die HKLF-Anweisung lautet also
HKLF 4 1 0 0 1 1 0 0 0 1 0
(5) Anschließend wird dieselbe Transformation auf die Gitterkonstanten angewendet. Aus
CELL 0.71069 9.046 10.268 16.899 90 90 90
ZERR 4 0.004 0.005 0.006 0 0 0
wird also:
CELL 0.71069 16.899 9.046 10.268 90 90 90
ZERR 4 0.006 0.004 0.005 0 0 0
(6) Für die anschließende Rechnung werden die Symmetrieanweisungen der Raumgruppe Pma2 eingefügt – fertig.
Etwas aufwendiger wird es, wenn Atomlagen berücksichtigt werden müssen. Man macht dann folgendes (die Symmetrieanweisungen der Nicht-Standardaufstellung wurden hier durch SIR eingefügt; es werden nur einige Atomlagen wiedergegeben:
TITL i012
CELL 0.71069 9.046 10.268 16.899 90 90 90
ZERR 4 0.004 0.005 0.006 0 0 0
LATT -1
SYMM -X,Y,-Z
SYMM -X,Y, 0.50000+Z
SYMM X,Y, 0.50000-Z
SFAC C H O NA RE N
UNIT 44 80 32 2 4 10
RE1 5 0.956787 -0.102886 0.250000 10.50000 0.02872 0.02288 =
0.04481 0.00000 0.00000 0.00412
C1 1 1.039637 -0.202516 0.178213 11.00000 0.05607 0.07380 =
0.09268 0.01567 -0.02027 0.01040
O1 3 1.089242 -0.273101 0.127311 11.00000 0.09327 0.06179 =
0.10259 0.01180 0.00381 0.01169
C2 1 0.790645 -0.217977 0.250000 10.50000 0.06614 0.02485 =
0.11512 0.00000 0.00000 -0.01295
O2 3 0.701140 -0.289079 0.250000 10.50000 0.03576 0.03625 =
0.11562 0.00000 0.00000 -0.02214
HKLF 4
Als erstes werden nun auf die beschriebene Art die HKLF-, die CELL- und die ZERR-Anweisung geändert. Als nächstes werden die SYMM-Anweisungen durch diejenigen der Standardaufstellung ersetzt. Zum Schluss werden die xyz-Werte der Atomlagen in der gleichen Weise behandelt wie die hkl-Werte und die Gitterkonstanten. Man könnte auch die anisotropen Temperaturfaktoren transformieren, die in der Reihenfolge U11, U22, U33, U23, U13, U12 angegeben sind, aber das ist viel vermeidbare Arbeit. Einfacher ist es, die jeweils zweite Zeile bei jedem Atom zu löschen, und außerdem in der ersten Zeile nur U11 stehen zu lassen. Bis hierhin sieht die Sache dann so aus:
TITL i012
CELL 0.71069 16.899 9.046 10.268 90 90 90
ZERR 4 0.006 0.004 0.005 0 0 0
LATT -1
SYMM -x, -y, z
SYMM x+.5, -y, z
SYMM .5-x, y, z
SFAC C H O NA RE N
UNIT 44 80 32 2 4 10
RE1 5 0.956787 -0.102886 0.250000 10.50000 0.02872
C1 1 1.039637 -0.202516 0.178213 11.00000 0.05607
O1 3 1.089242 -0.273101 0.127311 11.00000 0.09327
C2 1 0.790645 -0.217977 0.250000 10.50000 0.06614
O2 3 0.701140 -0.289079 0.250000 10.50000 0.03576
HKLF 4 1 0 0 1 1 0 0 0 1 0
Entsprechend der Transformation auf der HKLF-Anweisung wird nun auch bei xyz die Spalte mit den z-Werten ausgeschnitten und als erste Spalte mit Lageparametern wieder eingesetzt. Anschließend verfeinert man einige Zyklen isotrop, wobei unerwartet schlechte R-Werte anzeigen, dass bei einem der Schritte etwas schief gegangen ist. Wenn es geklappt hat, kann wieder alles anisotrop gesetzt werden und die Sache ist fertig.
| 
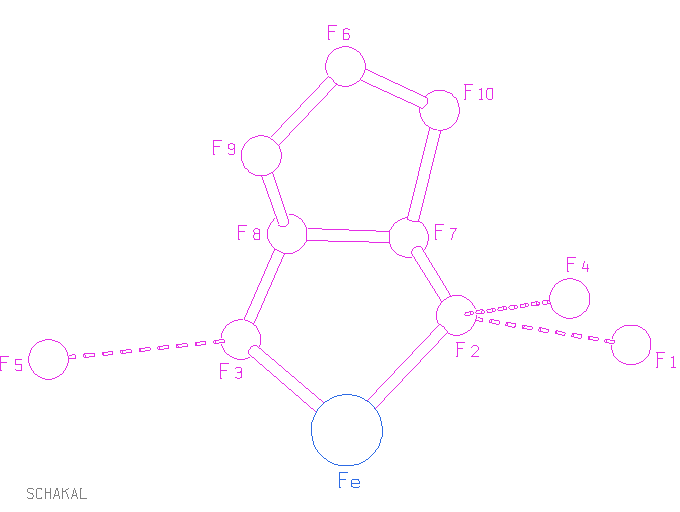{kind=link}
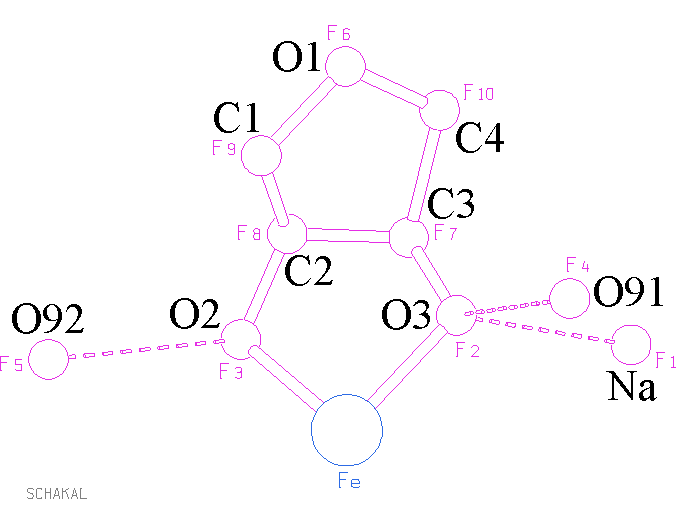{kind=link}
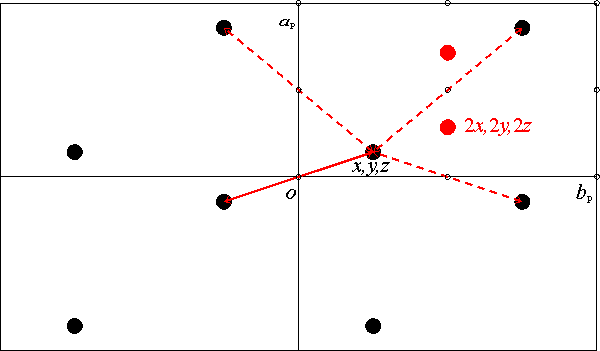{kind=link}